
 CC-BY 4.0
CC-BY 4.0
Introduction
Groundwater constitutes an extensive and globally prevalent ecosystem that contains the majority of accessible freshwater resources (Saccò et al., 2024). Animals inhabiting groundwater play a crucial role in enhancing the biodiversity of freshwater ecosystems and are essential for processes such as nutrient cycling and bioturbation (Bardgett & Van Der Putten, 2014). Groundwater offers numerous ecosystem services, ranging from supporting terrestrial and surface freshwater ecosystems to supplying drinking water (Griebler & Avramov, 2015). Therefore, investigating groundwater animals is vital for advancing our understanding of groundwater community structure and processes, ecosystem services, and their reactions to environmental changes (Maurice & Bloomfield, 2012).
With a remarkable diversity of over 430 identified species (Horton et al., 2023), the amphipod genus Niphargus Schiödte, 1849 is the most species-rich crustacean genus in subterranean waters. The distribution of this genus spans the Iberian Peninsula to the west and Iran to the east, covering European regions south of the limits of Quaternary glaciations (Stoch et al., 2024a). Studies on Niphargus are hampered by the lack of phylogenetic information on most species (except in a few well-studied areas) (Stoch et al., 2024a) and by the frequent occurrence of complexes of cryptic and pseudocryptic species Niphargus (Stoch et al., 2022). To date, studies on the phylogeny of the genus Niphargus have relied mainly on mitochondrial COI and nuclear 28S rRNA marker genes, and the resulting phylogenies remain poorly resolved, particularly for deeper nodes (Stoch et al., 2024b). Complete mitochondrial genome sequences of animals have been reported to yield well-resolved phylogenies (Cameron et al., 2007), but so far, not a single complete mitogenome sequence of Niphargus has been published.
To fill this gap and begin investigating the potential of niphargid complete mitochondrial genome sequences to shed new light on amphipod phylogenetic relationships, we focused on the Alpine species Niphargus dolenianensis (Lorenzi, 1898) and used genome skimming (Dodsworth, 2015) to assemble its complete mitogenome. Although the family Niphargidae is very diverse (over 230 described species following Horton et al., 2023), most species belong to the so-called Niphargus “megaclade” (Stoch et al., 2024a). Therefore, we selected one widely distributed species from the southern Alps (northern Italy) belonging to this megaclade for our study. We aimed to obtain a highly reliable Niphargus dolenianensis mitogenome for future comparative studies. For this purpose, we sequenced three different individuals using both nanopore and Illumina platforms and used Illumina reads to polish the assemblies of nanopore reads. Finally, to propose guidelines for future studies targeting niphargid mitogenomes, we tested whether Illumina-only and nanopore-only genome skimming would have been sufficient to obtain high-quality mitogenome sequences for downstream analysis.
Methods
DNA extraction and sequencing
Specimens of Niphargus dolenianensis (Lorenzi, 1898) (Fig.1) were collected in 2024 from two springs and a brook using a hand-net. Information on the specimens used in the analyses, collection sites, and DNA vouchers is presented in Table 1.

Figure 1 - Niphargus dolenianensis, habitus (a): male; (b): female.
Table 1 – List of specimens used in the study with the GenBank accession numbers of the deposited mitogenome sequences. All specimens were collected on 08/04/2024 by F. Stoch and G. Tomasin then identified morphologically. a.s.l. = above sea level.
Voucher | Accession number | Locality | Latitude (WGS84) | Longitude (WGS 84) | Altitude (m a.s.l.) |
|---|---|---|---|---|---|
FS_24.009 | PV534572 | San Giovanni al Natisone, spring near Villa Trento, Dolegnano, Italy | 13.420386 | 45.986732 | 80 |
FS_24.010 | PV534571 | Gorizia, spring along Vallone dell’Acqua road, Italy | 13.597421 | 45.954605 | 163 |
FS_24.011 | PV534570 | Gorizia, Cormons, Forest of Plessiva, springbrook, Italy | 13.488482 | 45.977869 | 97 |
Fresh samples were immediately preserved in 96% EtOH and then stored at -20°C at the Evolutionary Biology & Ecology unit of the Université libre de Bruxelles (ULB), Belgium. DNA extraction was performed from one or two pereopods (depending on the size of the specimen) using the Macherey-Nagel NucleoSpin Tissue kit, following the manufacturer’s protocol. The eluted DNAs was stored at -20°C. The three DNA extracts were sent for Illumina sequencing at the BRIGHTcore facility (Brussels, Belgium), enzymatic fragmentation was performed following by PCR-free library preparation and 2 x 151 bp paired-end sequencing on a Illumina NovaSeq 6000 machine. The same three DNA extracts were also used for long-read nanopore sequencing using Oxford Nanopore Technologies (ONT) with the Rapid PCR Barcoding kit SQK-RPB114.24, on a PromethION R10.4.1 flow cell.
The Illumina sequencing run cost us €8.35 (including VAT) per million reads, plus the cost for the library preparation that was around €80 (including VAT); from the time the DNA samples reached the laboratory, the turnaround time for obtaining raw data was four weeks (costs and processing times may vary among providers). For nanopore sequencing, the SQK-RPB114.24 kit (https://store.nanoporetech.com/rapid-pcr-barcoding-kit-24-v14.html) costs roughly €680 and supports 24 barcoded samples, with the kit usable for up to six runs, resulting in an estimated cost of €4.70 per sample. To be added is the cost of PromethION flow cell: the prize of a new flow cell is currently €1083 (including VAT) when ordered by pack of four; each flow cell can support two runs of 24 mitochondrial genomes each, hence a cost of €22.5 per sample.
Preparation of the nanopore library required about 15 min, in addition to the PCR that took a total of 1 hour and 40 minutes with the following conditions: 3min at 95°C; 14 cycles of 15sec at 95°C, 15sec at 56°C, 6min at 65°C; 6min at 65°C, then hold 10°C. Raw sequencing data were available within 24 h after loading the flow cell.
Mitochondrial genome annotation and analysis
Illumina data were trimmed using Skewer (Jiang et al., 2014) in paired-end mode. Nanopore data were basecalled using Guppy v3.8 (in super-high-quality mode) to generate fastq reads from the raw fast5. Nanopore reads were filtered to retain only reads with an average quality score of at least 14 using the script split_qscore.py available as part of the Buttery-eel package (Samarakoon et al., 2023) and assembled using Flye v2.9.6 (Kolmogorov et al., 2019), as well as with hifiasm v0.25.0-r726 (Cheng et al., 2021) for comparison. The resulting graphical fragment assembly (GFA) files were examined using Bandage (Wick et al., 2015) to identify and extract the circular contig of the mitochondrial genome. The first automatic Illumina polishing of the nanopore-assembled mitochondrial contig was performed using Polypolish (Wick & Holt, 2022). The resulting assemblies were checked closely for structural and base-level errors by mapping the nanopore and Illumina reads on them using minimap2 (Li, 2018), converting the resulting SAM file into BAM, sorting it using SAMtools (Li et al., 2009), and visualizing the resulting read pileups using Tablet (Milne et al., 2010). The few remaining errors detected in Tablet were corrected manually in the contig FASTA files using AliView (Larsson, 2014). For comparison, Illumina-only assemblies of the three mitochondrial genomes were attempted using the NOVOplasty assembler (Dierckxsens et al., 2017), using as seed the previously published COI sequence (Genbank accession KY706720) of a Niphargus dolenianensis individual (Eme et al., 2018).
A quick annotation of each nanopore-assembled, Illumina-polished mitogenome sequence was performed using GeSeq (Tillich et al., 2017), with tRNAscan-SE v2.0.7 (Chan et al., 2021) to locate tRNAs; we used the available mitogenomes of Pseudoniphargus stocki and Pseudoniphargus carpalis (Stokkan et al., 2018) as “3rd Party References”. A second annotation was conducted using MITOS2 (Bernt et al., 2013), specifying the invertebrate mitochondrial genetic code 5 and the reference dataset RefSeq63 Metazoa.
Considering that no Niphargus genome was available in GenBank, we manually checked and refined all annotations, including the possible presence of stop codons of the protein-coding genes and the predicted secondary structure of tRNA genes (using the RNAfold tool available on the ViennaRNA web service; Gruber et al., 2015). To detect initial and terminal codons (stop codon or part of it, such as T– or TA–), sequences were aligned with reference sequences of a very closely related genus (Pseudoniphargus: Weber et al., 2021), considering that their sequences can overlap with tRNA sequences (Stokkan et al., 2016). The resulting annotated sequences are available in GenBank (accession numbers PV534570, PV534571 and PV534572).
The program mtSVG (available at https://github.com/odethier-ulb/mtSVG) was used to create a visual summary of the final annotated mitogenome of N. dolenianensis based on the three mitogenome sequences obtained in the present study. Read coverage depth graphs were created from BAM files using Grace-5.1.17 (Vaught, 1996). Nucleotide diversity analyses of the 13 protein-coding genes and two ribosomal RNA genes were conducted using the packages pegas (Paradis, 2010) and seqinR (Charif & Lobry, 2007) in RStudio 4.4.1.
Phylogenetic reconstruction
Together with our three Niphargidae mitogenome sequences, we included a selection of representative complete mitogenomes from the amphipod families Pseudoniphargidae (Stokkan et al., 2018), Gammaridae (Macher et al., 2017; Cormier et al., 2018; Mamos et al., 2021), Metacrangonyctidae (Bauzà-Ribot et al., 2009; Bauzà-Ribot et al., 2012), Crangonyctidae (Benito et al., 2021), Talitridae (Kumar Patra et al., 2019), and Hyalellidae, as well as two isopod species (Kilpert & Podsiadlowski, 2006; Kilpert et al., 2012), as an outgroup (all GenBank accession numbers are available in Table 2). Families were selected based on their putative affinity with Niphargidae following Copilaş-Ciocianu et al. (2020). Unfortunately, the mitogenome sequences for amphipods are limited to a few families; the goal of building this tree was therefore only to allocate the niphargids in an updated, mitogenome-based phylogeny to identify its sister family.
The translated protein sequences of all protein-coding genes were concatenated and used to build a maximum-likelihood (ML) phylogenetic tree using the program IQ-TREE2 (Nguyen et al., 2015) with the invertebrate mitochondrial amino acids substitution model (MtInv) (Le et al., 2017), and node support was assessed using 50,000 ultrafast bootstrap replicates (Hoang et al., 2018). The evolutionary model employed (MtInv) is the usual one implemented in IQ-TREE2 for amino acids and the same used by Macher et al. (2023) in a previous study of amphipod metagenomes. According to Macher et al. (2023), mitochondrial ribosomal genes are much more difficult to align than protein-coding genes; therefore, rDNA genes (rrnL and rrnS) were not included in the analysis. A linear representation of the order of protein-coding genes in each mitogenome was generated using the program mtSVG and these images were added next to the leaves of the ML phylogenetic tree.
Table 2 – List of all the complete mitochondrial sequences downloaded from GenBank used for mitochondrial genome analysis and integrated with new ones used to build the ML tree.
Species: | GenBank code: |
|---|---|
Asellus aquaticus | GU130252 |
Echinogammarus berilloni | BK059223 |
Gammarus fossarum | NC_034937 |
Gammarus lacustris | NC_044469 |
Gammarus roeselii | NC_037481 |
Hyalella azteca | MH542433 |
Ligia oceanica | DQ442914 |
Marinogammarus marinus | BK059224 |
Metacrangonyx dominicanus | HE860499 |
Metacrangonyx ilvanus | NC_019656 |
Metacrangonyx longipes | NC_013032 |
Metacrangonyx repens | HE860495 |
Pectenogammarus veneris | BK059233 |
Pseudoniphargus grandis | MH592128 |
Pseudoniphargus morenoi | MH592132 |
Pseudoniphargus sp. 2-Canaries | MH592142 |
Pseudoniphargus stocki | NC_039354 |
Stygobromus allegheniensis | NC_046511 |
Stygobromus indentatus | NC_030261 |
Stygobromus pizzinii | NC_046510 |
Stygobromus tenuis potomacus | KU869712 |
Trinorchestia longiramus | MH542431 |
Results
The final mitochondrial genome sequences of the three Niphargus dolenianensis individuals ranged from 14,964 to 15,097 bp in length (Fig.2). The details of their annotation are reported in Table 3. Their global GC content was 24.6% for FS_24.009, 23.5% for FS_24.010 and 23.0% for FS_24.011.
For the sample FS_24.009 we obtained from Illumina and nanopore, respectively, 2.82 and 1.71 Gbp, for FS_24.010, 2.66 and 0.69 Gbp, and for FS_24.011, 0.95 and 1.70 Gbp. The coverage depth obtained for the assembled mitogenomes generated in Illumina short reads ranged from approximately 40 to 250X; similarly, the coverage depth for nanopore long reads ranged from 40 to 300X (Fig.5). Nanopore coverage depth profiles were more uneven than those for Illumina, possibly because of biases caused by the PCR amplification step in the Rapid PCR Barcoding Kit protocol.
The sequences and predicted secondary structures of the tRNA genes are shown in Fig.3. Only tRNA-Gly and tRNA-Leu2 were identical across all three individuals, whereas all other tRNA sequences had a few base differences (highlighted in pink in Fig.3). These differences were mostly substitutions (most often located outside of stem structures, except for a few mutations in stems that either did not disturb base pairing or, in the case of tRNA-Met, that was compensated by a mutation in the facing position). Of the 22 tRNAs annotated in the mitochondrial genome of Niphargus dolenianensis, 19 had the typical 3-arm structure, whereas tRNA-Ser1 and tRNA-Val lacked the dihydrouridine (DHU) arm and tRNA-Phe lacked the T-arm. The structure of tRNA-Val of FS_24.011 could not be predicted de novo from its sequence using the Vienna server because of the extra G-A pairing of this sequence compared with the other two. Therefore, we constrained its folding to the structure shown in Fig.3 using the results obtained from the two other individuals.
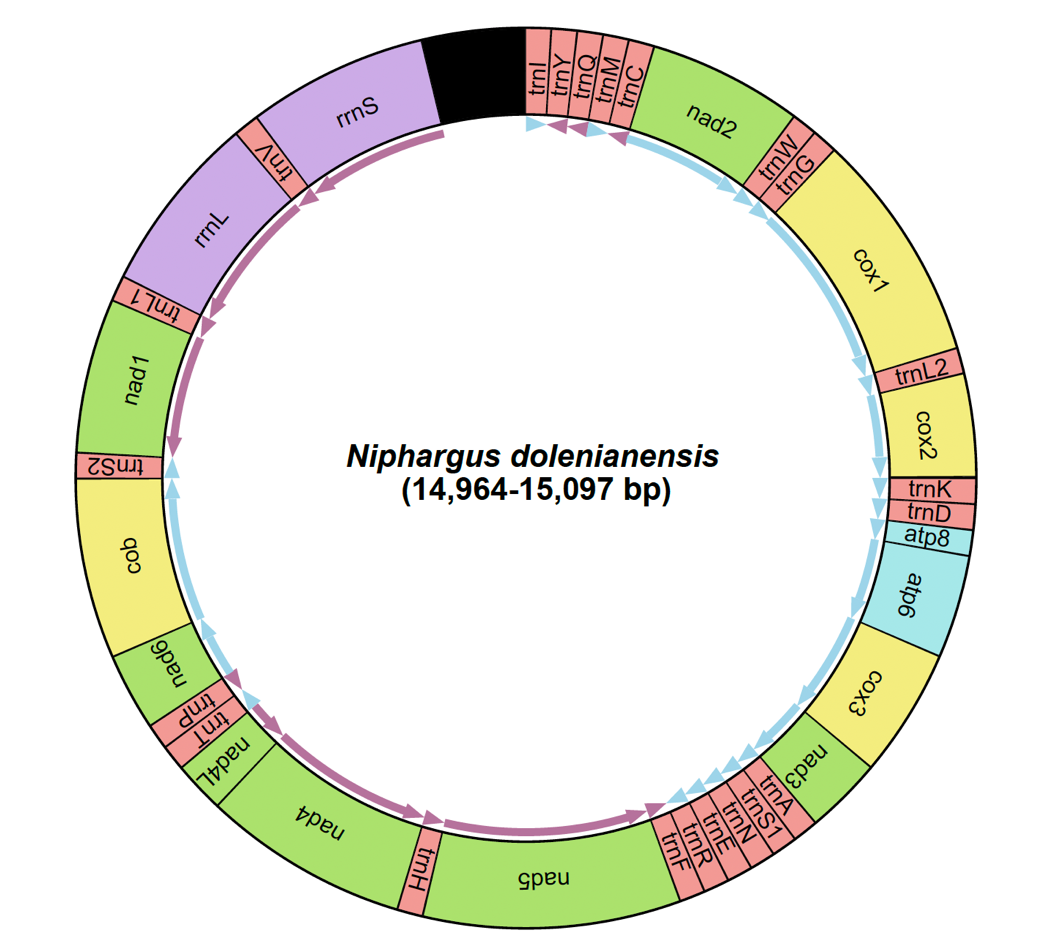
Figure 2 – Organization of the mitochondrial genome of Niphargus dolenianensis showing the arrangement of the 13 protein-coding genes, the twelve tRNAs, the two rRNA genes and the putative control region (in black).
The nucleotide diversity (\(\pi\)) values for each protein-coding and ribosomal RNA gene are shown in Fig.4. The values ranged from 0.06 to 0.09 for the protein-coding genes, whereas it was only 0.03-0.04 for the two ribosomal RNA genes.
There were no significant differences between the Flye and Hifiasm mitogenome assemblies obtained from the nanopore long reads: for FS_24.009, the two were perfectly identical, whereas in the case of FS_24.010 and FS_24.011, there were only two differences between the Flye and Hifiasm mitogenome assemblies, in both short indels at the level of long homopolymers. These indels and several others (always located in long homopolymers) in the nanopore-assembled sequences were corrected through polishing using Illumina reads. In the mitogenome of FS_24.009, the differences between the Flye nanopore assembly before and after Illumina polishing were: a one-base indel in nad4L and a three-base indel in rrnS; in the mitogenome of FS_24.010, a one-base indel in atp6, another one in nad4L, and a two-base indel in rrnS; in the mitogenome of FS_24.011, a one-base indel in nad4L, and a two-base indel in rrnS.
Table 3 – Position of all coding and non-coding genes, number of amino acids in PCGs and their start and stop codons. The underlined numbers represent the position where genes overlap.
Region | Position | FS_24.009 AA Start | Stop | Position | FS_24.010 AA Start | Stop | Position | FS_24.011 AA Start | Stop | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
trnI | 1-61 | 1-61 | 1-60 | |||||||||
trnY | 64-122 | 64-122 | 113-171 | |||||||||
trnQ | 119-176 | 119-176 | 168-225 | |||||||||
trnM | 227-287 | 274-334 | 277-337 | |||||||||
trnC | 288-346 | 335-393 | 338-396 | |||||||||
nad2 | 348-1338 | 330 | ATT | T— | 395-1385 | 330 | ATA | T— | 398-1388 | 330 | ATT | T— |
trnW | 1339-1401 | 1386-1448 | 1389-1451 | |||||||||
trnG | 1406-1469 | 1452-1515 | 1455-1518 | |||||||||
cox1 | 1470-3006 | 512 | ATA | T— | 1516-3052 | 512 | ATA | T— | 1519-3055 | 511 | ATT | T— |
trnL2 | 3007-3068 | 3053-3114 | 3056-3117 | |||||||||
cox2 | 3069-3741 | 224 | ATA | T— | 3115-3787 | 224 | ATA | T— | 3118-3790 | 224 | ATA | T— |
trnK | 3742-3802 | 3788-3848 | 3791-3851 | |||||||||
trnD | 3803-3862 | 3849-3908 | 3852-3911 | |||||||||
atp8 | 3863-4024 | 53 | ATT | TAA | 3909-4070 | 53 | ATT | TAA | 3912-4073 | 53 | ATC | TAA |
atp6 | 4018-4686 | 222 | ATG | TAA | 4064-4732 | 222 | ATG | TAA | 4067-4735 | 222 | ATG | TAA |
cox3 | 4687-5475 | 262 | ATG | TAA | 4733-5524 | 263 | ATG | TAA | 4736-5524 | 262 | ATG | TAA |
nad3 | 5484-5837 | 117 | ATG | TAG | 5530-5883 | 117 | ATG | TAG | 5533-5886 | 117 | ATG | TAG |
trnA | 5836-5896 | 5882-5942 | 5885-5945 | |||||||||
trnS1 | 5896-5947 | 5942-5993 | 5945-5996 | |||||||||
trnN | 5947-6008 | 5993-6054 | 5996-6057 | |||||||||
trnE | 6006-6069 | 6052-6115 | 6055-6118 | |||||||||
trnR | 6064-6122 | 6110-6168 | 6113-6171 | |||||||||
trnF | 6121-6180 | 6167-6225 | 6170-6229 | |||||||||
nad5 | 6181-7888 | 569 | ATA | T— | 6226-7933 | 569 | ATA | T— | 6230-7937 | 569 | ATA | T— |
trnH | 7889-7948 | 7934-7993 | 7938-7997 | |||||||||
nad4 | 7949-9263 | 438 | ATG | T— | 7994-9308 | 439 | ATG | T— | 7998-9312 | 438 | ATG | T— |
nad4L | 9257-9547 | 96 | ATG | TAA | 9302-9592 | 96 | ATG | TAA | 9306-9596 | 96 | ATG | TAA |
trnT | 9551-9610 | 9595-9654 | 9599-9658 | |||||||||
trnP | 9609-9670 | 9653-9714 | 9657-9718 | |||||||||
nad6 | 9673-10170 | 165 | ATG | TAA | 9729-10214 | 161 | ATT | TAA | 9733-10218 | 161 | ATT | TAA |
cob | 10170-11309 | 379 | ATG | TAA | 10214-11353 | 379 | ATG | TAA | 10218-11357 | 379 | ATG | TAA |
trnS2 | 11308-11367 | 11352-11411 | 11356-11415 | |||||||||
nad1 | 11365-12276 | 303 | ATT | TAA | 11409-12320 | 303 | ATT | TAA | 11413-12324 | 303 | ATT | TAA |
trnL1 | 12286-12347 | 12330-12391 | 12334-12395 | |||||||||
rrnL | 12325-13383 | 12369-13427 | 12373-13432 | |||||||||
trnV | 13384-13429 | 13428-13473 | 13433-13478 | |||||||||
rrnS | 13430-14487 | 13474-14528 | 13479-14531 | |||||||||
CR | 14488-15097 | 14529-14964 | 14532-15069 | |||||||||
By contrast, the Illumina-only assemblies obtained using NOVOplasty had many discrepancies with the nanopore-assembled, Illumina-polished accurate sequences: the majority of the differences were detected in the control regions with various insertions, and few indels and base substitutions were also found in tRNAs as well as in some protein-coding genes. For the individual FS_24.009, we detected four substitutions in trnY and one substitution in both nad3 and nad4L; for the individual FS_24.010, we detected four substitutions in trnY, a 43-base deletion between trnQ and trnM, and one-base substitution in nad3; for the individual FS_24.011, we detected four substitutions in trnY, a 55-base deletion between trnI and trnY, a 5-base deletion between trnG, and one substitution in nad3.
The ML tree obtained from this analysis is shown in Fig.6. The newly annotated mitogenome of Niphargus formed a clade with Pseudoniphargus, supported by a 99% ultrafast bootstrap value. No difference was found in the protein-coding gene order between Niphargidae and Pseudoniphargidae, but some differences were detected between more distant amphipod families. Most Gammaridae and all Crangonyctidae had the same gene order as Niphargidae and Pseudoniphargidae, with the exception of Marinogammarus marinus for which nad5, rrnS and rrnL were translocated. In the case of Metacrangonyctidae, cytb changed position and orientation; whereas in Hyalellidae, nad1 changed orientation but remained in the same location. The talitrid Trinorchestia longiramus had nad3 located before cox1 and its cytb and nad6 were swapped.
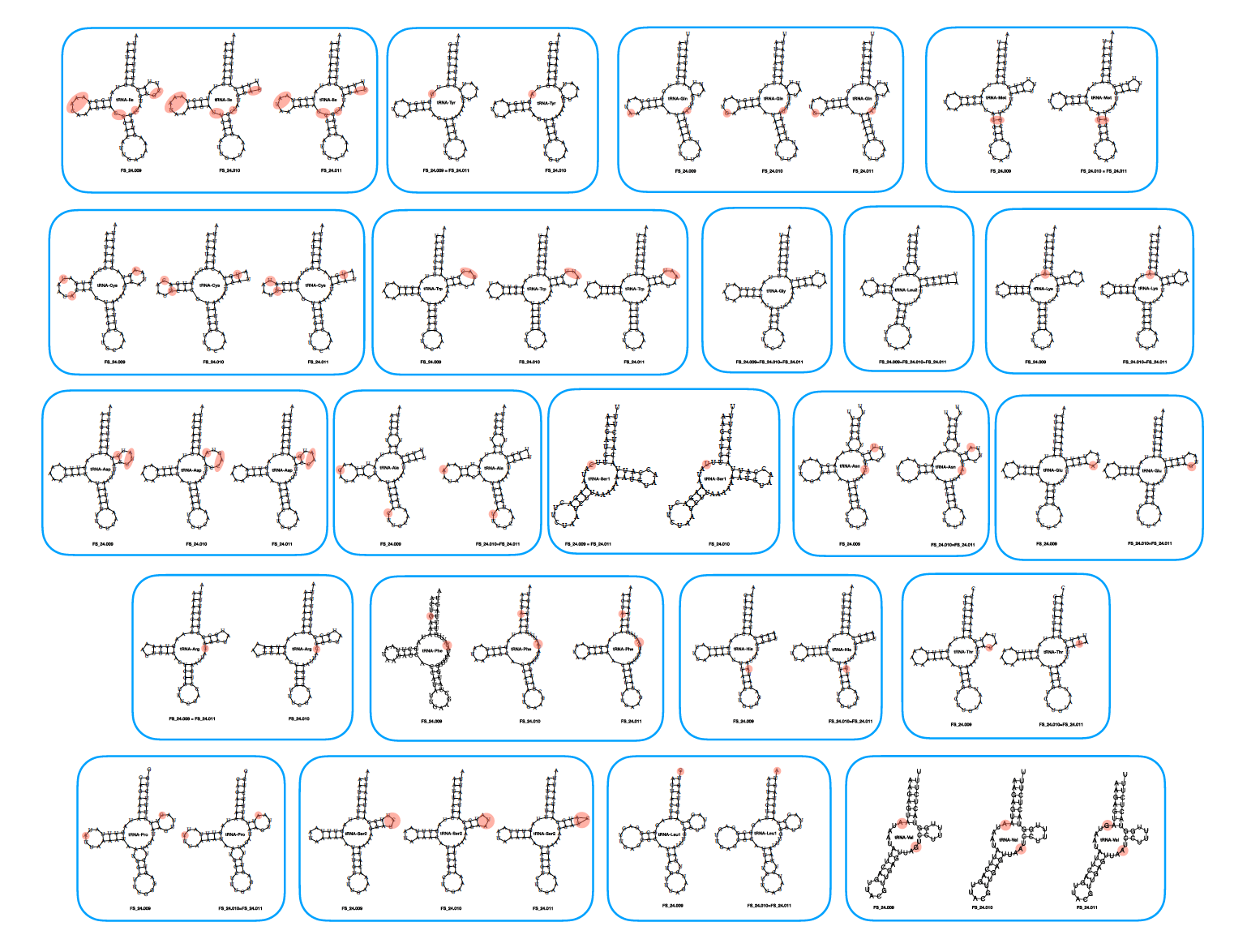
Figure 3 – Predicted secondary structure of the 22 tRNA genes.
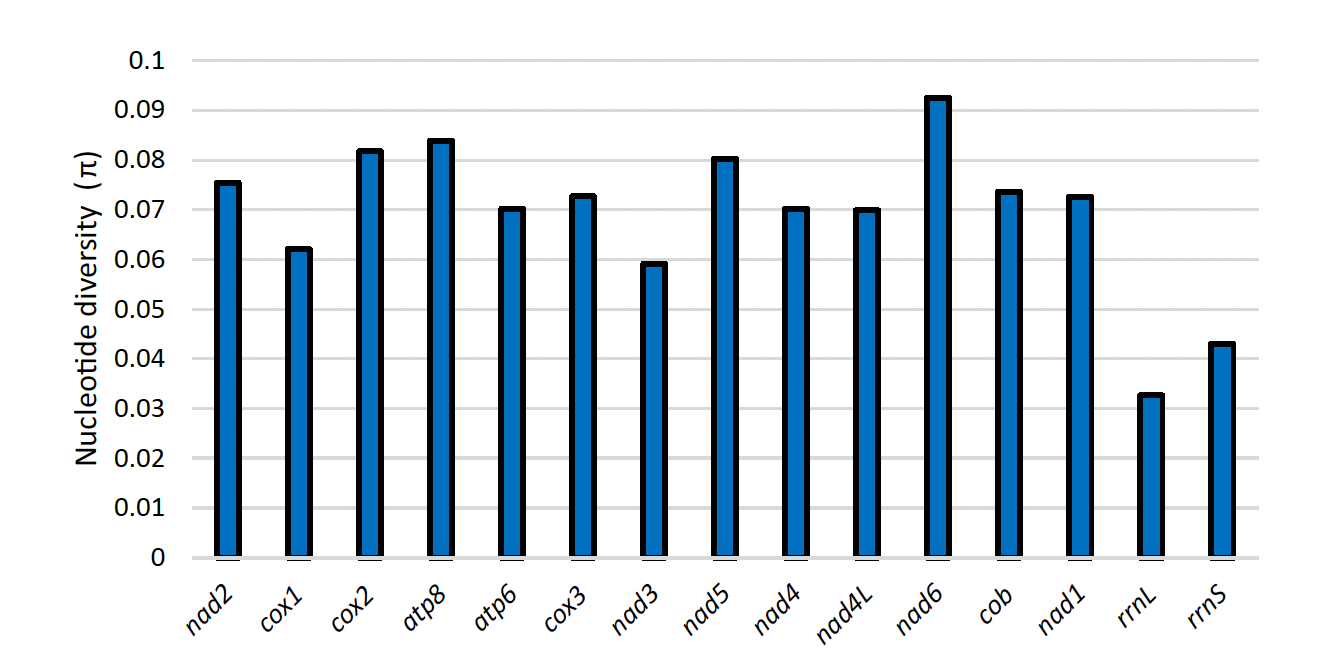
Figure 4 – Nucleotide diversity of the three Niphargus mitogenomes for protein-coding genes and rRNA genes.
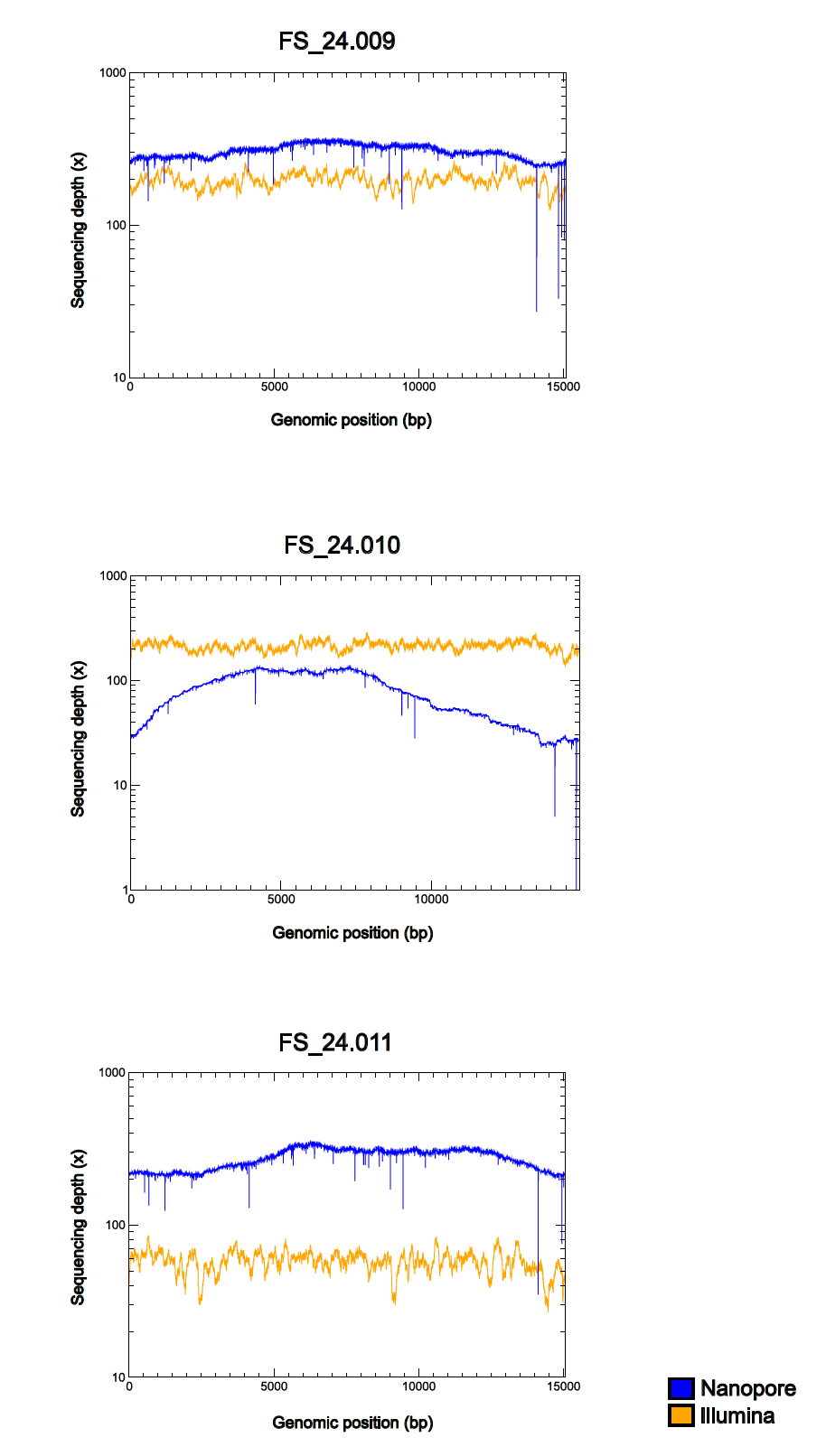
Figure 5 – Read coverage graph comparing Illumina and nanopore data for the three Niphargus individual. Graphs with logarithmic y-axis.
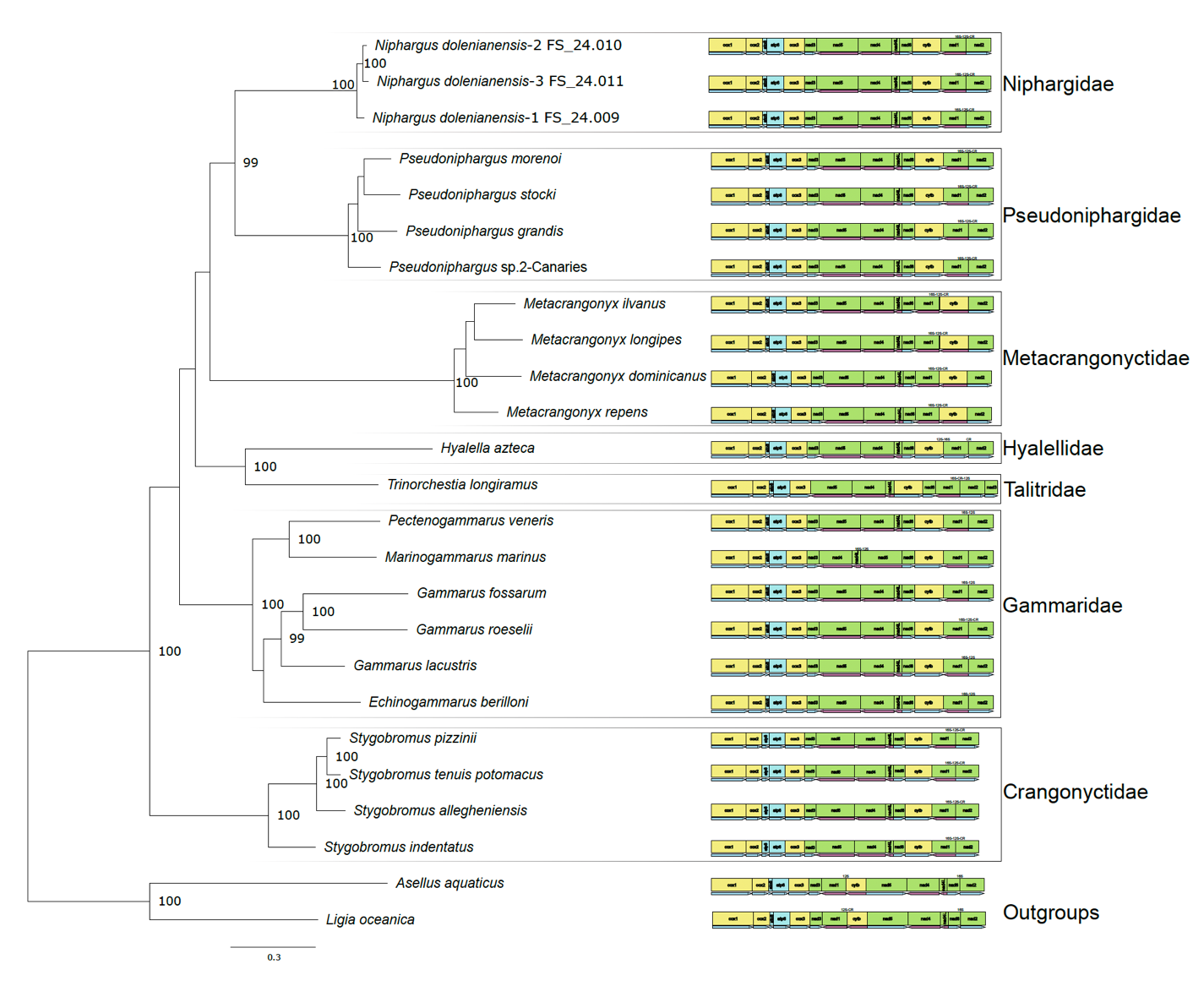
Figure 6 – Maximum-likelihood phylogenetic tree obtained from translated protein sequences of all mitochondrial protein-coding genes. Different families are indicated on the right side. Labels of nodes with a probability below 95 % were removed. 16S, 12S and CR represent the position of the rrnL and rrnS genes as well as the control region, when available from the annotation on Genbank.
Discussion
Like other metazoans, the mitochondrial genome of crustaceans typically consists of a circular double-stranded DNA molecule ranging from 12 to 20 kb, with a highly conserved gene content. It contains 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs), and a large non-coding region where replication of the mitochondrial genome is initiated, called the D-loop or control region (CR) (Boore, 1999).
In this study, we present the results obtained from the first three complete mitogenomes of the genus Niphargus, all belonging to the species N. dolenianensis. Nanopore assembly followed by careful Illumina polishing (first automatically, then manually) yielded genome annotations that were highly consistent with published amphipod mitogenomes, particularly with those of Pseudoniphargidae, which, based on our phylogenetic reconstruction using protein-coding genes, is the sister family to Niphargidae, as already hypothesized by Weber et al. (2021).
Most mt-tRNAs fold into the same cloverleaf secondary structure as nuclear-encoded tRNA sequences, comprising four stems and three loops (Jühling et al., 2012). However, as reported in the genus Pseudoniphargus (Stokkan et al., 2016), the tRNA-Ser1 and tRNA-Val of Niphargus dolenianensis lacked the dihydrouridine (DHU) arm, also common in nearly all metazoans (Ki et al., 2010), while its tRNA-Phe lacked the T-arm. The tRNA secondary structures generally contained wobble base pairs (G–U, I–U, I–A, and I–C); however, we also noticed some non-canonical pairing (e.g. U–U, G–G, U–C, A–A), as frequently observed in other species (Ki et al., 2010). Nucleotide diversity values were higher for protein-coding genes than for ribosomal RNA genes. Among the protein-coding genes, a higher diversity value was observed for nad6, whereas cox1 had a comparatively lower value, even though the latter is commonly used to identify arthropod species.
Using nanopore for genome skimming allowed us to exclude nuclear mitochondrial DNA (NUMT) segments because the length of nanopore reads is larger than the typical length of NUMTs (Richly, 2004). Illumina-only assemblies were imperfect, with many structural errors (mainly in the control region and in ribosomal RNA genes, although adjacent tRNA regions were also affected), probably due to short-read assembly, whereas nanopore-only assemblies were plagued by one-based artifactual indels in coding regions (caused by homopolymers longer than 10-12 bases), resulting in reading-frame shifts. However, despite Illumina sequencing providing a higher amount of data (Gbp) per sample (except for FS_24.011, for which the nanopore-only assembly generated more data), this increase did not translate into improvements in either the final assembly quality or coverage depth compared to the nanopore-only assembly.
The fact that the nanopore-only assembly contained some artifactual indels was unexpected, given that nanopore R10.4.1 data have been reported to yield error-free bacterial genome sequences that do not require Illumina polishing (Sereika et al., 2022). As mitochondria are alpha-proteobacteria (Fan et al., 2020), the same approach would have been expected to work on mitogenomes as well. However, the mitogenome of Niphargus dolenianensis contains many long stretches of polyA and polyT (up to a length of 25 identical nucleotides in a row) that are not typically observed in bacterial genomes and are responsible for the artifactual indels observed. These stretches were mainly observed in the two rDNA genes and in the control region, although in some cases they disrupted protein-coding genes. These errors were manually corrected after checking amino acids translation, Illumina outputs and nanopore-based sequences of the three different. This is most probably caused by the extremely low GC content of the mitochondrial genome of Niphargus dolenianensis, which, being lower than 25% GC, falls outside the range of GC content of bacteria and archaea, except for a very few cases such as Carsonella (Mann & Chen, 2010).
In such cases, both nanopore and Illumina data are required to obtain a highly accurate, error-free mitogenome sequence. This is particularly important when producing the first reference mitogenome sequence for a previously unexplored genus or family, as was the case here. In the future, resequencing of other species closely related to the one for which a reference sequence was generated could rely on nanopore only, since all the artifactual errors in the resulting genome assembly will be located at the level of long homopolymers and will be easily detected and corrected by hand. In terms of both cost and turnaround time, nanopore sequencing is markedly faster and more economical, with the potential to generate final raw data in less than two days from the initial processing of animal tissue. Sequencing the mitogenomes of three closely related specimens, as we did, allowed us to cross-compare and refine our gene annotations and was also instrumental in obtaining properly folded sequences for all tRNAs, as the tRNA-Val of FS_24.011 could not be folded properly without relying on information from the other two. Therefore, if we had only sequenced FS_24.011, we could have incorrectly concluded that tRNA-Val was absent in Niphargus dolenianensis.
The phylogenetic tree of amphipods, using the complete set of protein-coding genes in the mitochondrial genome, yielded a well-resolved phylogeny and confirmed the close evolutionary relationship between Niphargidae and Pseudoniphargidae, which formed together a clade supported by very strong bootstrap values. This highlights the usefulness of complete mitochondrial genome sequences for conducting studies at a deeper phylogenetic level (Bauzà-Ribot et al., 2009; Sun et al., 2018), with the known limitations of mitochondrial genomes, mainly due to saturation problems, in resolving some ancient splits that are millions of years old (Phillips et al., 2013). It is well known that the saturation problem of the cox1 gene in amphipods (Stoch et al., 2022; Stoch et al., 2024a) leads to poor resolution of ancient splits. The phylogenetic tree generated using complete mitogenome sequences of amphipods made it possible to resolve some of these basal nodes (Bauzà-Ribot et al., 2013; Stokkan et al., 2018; Höpel et al., 2022). Our research offers valuable insights into nanopore sequencing that can be employed to obtain more precise mtDNA genome sequences, which is promising for gaining a clearer understanding of the evolution and diversification of amphipods.
Acknowledgements
Preprint version 2 of this article was peer-reviewed and recommended by Peer Community In Zoology (Schon, 2025: https://doi.org/10.24072/pci.zool.100370).
The authors wish to thank Laurent Grumiau and Florence Rodriguez Gaudray for their assistance with the laboratory work.
We sincerely thank Miquel Arnedo and Gontran Sonet for their constructive reviews and valuable suggestions, which greatly improved the quality of this manuscript. We also express our gratitude to Isa Schon for handling the evaluation of our article as Recommender for PCI Zoology.
Author contributions
AS conducted most of the laboratory work, contributed to data analysis and interpretation, and drafted the manuscript with input from all authors. NL contributed to the laboratory work. FS collected all samples and contributed to data analysis, mitogenome annotation, and interpretation. JFF conducted most of the data analyses and contributed to their interpretation and validation. All authors reviewed and approved the final manuscript.
Data, scripts, code, and supplementary information availability
Assembly of nanopore reads using Flye:
mkdir Q14min
gzip -c -d *.fastq.gz|split_qscore.py -q 14 -f fastq - Q14min
cd Q14min/
rm reads.fail.fastq
flye—nano-hq reads.pass.fastq—threads 10 -o flye
Assembly of nanopore reads using hifiasm:
hifiasm—ont reads.pass.fastq -t10 -o hifiasm
Checking the nanopore coverage:
minimap2 -a -t 10 -x map-ont mtgenome.fasta reads.fastq > checknanopore.sam
samtools view -b checknanopore.sam -o checknanopore.bam
samtools sort checknanopore.bam -o checknanopore_sorted.bam
samtools index checknanopore_sorted.bam
Comparison with Illumina assembly and polishing:
skewer -m pe R1_001.fastq.gz “R2_001.fastq.gz
mv R1_001.fastq-trimmed-pair1.fastq R1trimmed.fastq
mv “R1_001.fastq-trimmed-pair2.fastq R2trimmed.fastq
micromamba activate polypolish
module load BWA
bwa index mtDNA_circular.fasta
bwa mem -t 16 mtDNA_circular.fasta R1trimmed.fastq > R1trimmed.sam
bwa mem -t 16 _mtDNA_circular.fasta R2trimmed.fastq > R2trimmed.sam
polypolish filter—in1 R1trimmed.sam—in2 R2trimmed.sam \
--out1 R1trimmed_filtered.sam—out2 R2trimmed_filtered.sam
polypolish polish mtDNA_circular.fasta R1trimmed_filtered.sam \
R2trimmed_filtered.sam > mtDNA_circular_polished.fasta
Plotting a circular map of the genome:
mtSVG.py—gff FS\_24.010.gff3 --size 14964 --species “Niphargus dolenianensis”—circular—start trnI—oriented—intergenic 100
Plotting coverage graphs:
genomeCoverageBed -ibam checknanopore_sorted.bam -d > checknanopore.cov
genomeCoverageBed -ibam checkIllumina_sorted.bam -d > checkIllumina.cov
cut -f 2,3 checkIllumina.cov|xmgrace -
cut -f 2,3 checknanopore.cov|xmgrace -
cut -f 2,3 checknanopore.cov > nanopore.cov
cut -f 2,3 checkIllumina.cov > Illumina.cov
xmgrace Illumina.cov nanopore.cov
Nucleotide diversity using Rstudio 4.4.1:
library(pegas)
library(seqinr)
file_paths <- c(“atp6.fasta”,
”atp8.fasta”,
”cob.fasta”,
”cox1.fasta”,
”cox2.fasta”,
”cox3.fasta”,
”nad1.fasta”,
”nad2.fasta”,
”nad3.fasta”,
”nad4L.fasta”,
”nad4.fasta”,
”nad5.fasta”,
”nad6.fasta”,
”rrnL.fasta”,
”rrnS.fasta”)
nuc_div_results <- list()
for (file in file_paths) {
# Read the gene sequence
gene_data <- read.dna(file, format = “fasta”)
# Calculate nucleotide diversity
nuc_div_result <- nuc.div(gene_data)
# Store the result with the file name (gene name)
gene_name <- basename(file) # Extract the file name to use as the gene name
nuc_div_results[[gene_name]] <- nuc_div_result
}
print(nuc_div_results)
write.csv(nuc_div_df, “C:/Users/Alice/Desktop/nucleotide_diversity_results.csv”, row.names = FALSE)
nuc_div_df <- data.frame(Gene = character(), Nucleotide_Diversity = numeric(), stringsAsFactors = FALSE)
for (file in file_paths) {
gene_data <- read.dna(file, format = “fasta”)
nuc_div_result <- nuc.div(gene_data)
gene_name <- basename(file)
ML phylogenetic tree using IQtree2:
iqtree2 -s Mitogenomes_PCGs.phy -m mtInv+I+G4 -nt auto -B 50000
Conflict of interest disclosure
The authors declare no financial or personal interest that could appear to have influenced the work reported in this study.
Funding
AS’s Ph.D. was supported by a one-year Université libre de Bruxelles (ULB) seed grant and subsequently by DarCo (The vertical dimension of conservation: a cost-effective plan to incorporate subterranean ecosystems in post-2020 biodiversity and climate change agendas, BIODIV21_0006). Her Ph.D. was also supported by a prize awarded by the David and Alice Van Buuren Fund and the Jaumotte-Demoulin Foundation.
NL’s Ph.D. was supported by a seed grant for the ULB-VUB collaborative research. FS and JFF were supported by Projet de Recherches’ grant no. T.0078.23 to JFF.
This study is part of the DarCo project funded by Biodiversa+, the European Biodiversity Partnership under the 2021–2022 BiodivProtect joint call for research proposals, co-funded by the European Commission (GA no. 101052342) and the Ministry of Universities and Research (Italy), Agencia Estatal de Investigación—Fundación Biodiversidad (Spain), Fundo Regional para a Ciência e Tecnologia (Portugal), Suomen Akatemia—Ministry of the Environment (Finland), Belgian Science Policy Office (Belgium), Agence Nationale de la Recherche (France), Deutsche Forschungsgemeinschaft e.V. (Germany), Schweizerischer Nationalfonds (grant no. 31BD30_209583, Switzerland), Fonds zur Förderung der Wissenschaftlichen Forschung (Austria), Ministry of Higher Education, Science and Innovation (Slovenia), and the Executive Agency for Higher Education, Research, Development and Innovation Funding (Romania).