Latest Articles
-
Section: Plants ; Topics: Agricultural sciences
Enhancing sugarcane yield resilience under a changing climate: adaptive irrigation and varietal strategies in Reunion Island
Christina, Mathias; Pilloni, Raphaël;
Mézino, Mickaël;
Loison, Romain;
Le Mézo, Lionel;
Poser, Christophe
Pilloni, Raphaël;
Mézino, Mickaël;
Loison, Romain;
Le Mézo, Lionel;
Poser, Christophe
10.24072/pcjournal.764 - Peer Community Journal, Volume 6 (2026), article no. e75
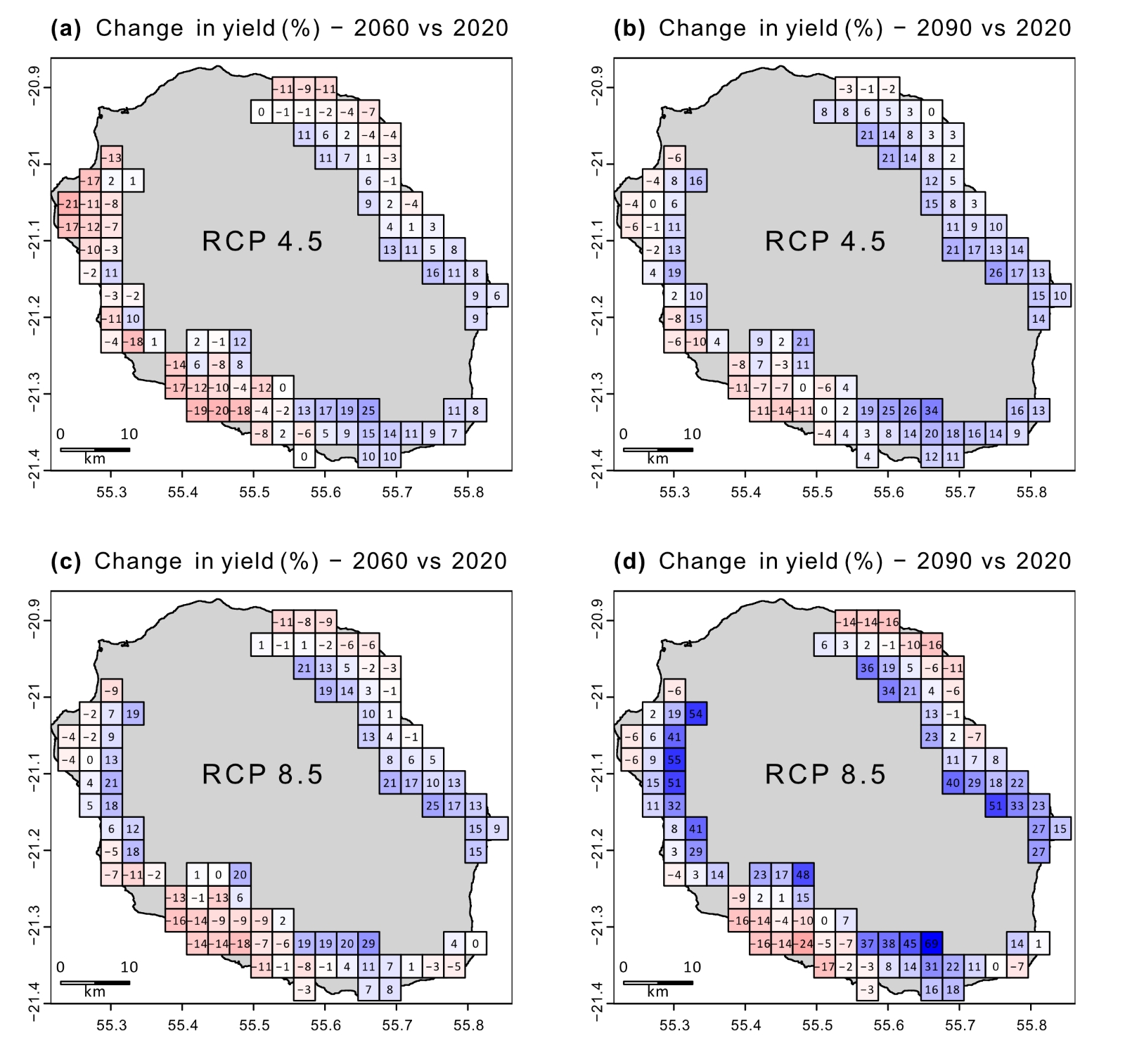
Get full text PDFVariations in rainfall patterns, whether in quantity or frequency, pose a significant challenge that sugarcane crops must adapt to in the context of climate change. This study aimed to assess the impact of future climate scenarios from 2015 to 2100 on sugarcane yield at a regional scale in Reunion Island and to identify adaptation practices to mitigate the impact of climate. Different irrigation regimes were tested to assess the impact on growth and future water requirements using the Mosicas sugarcane crop growth model at a 3 x 3 km resolution. High-resolution climate data for three IPCC future scenarios (RCP 2.5, 4.5, and 8.5) were obtained from the BRIO project, conducted by Météo France, the French national weather service. Results showed that, despite a favorable increase in temperature, changes in precipitation patterns resulted in minimal yield change at the island scale compared to the current period. However, the impact of climate change varied significantly across different climatic areas on the island. On the south-west coast, yields decreased significantly (by -8% and -4% on average in 2060 and 2090, respectively), while on the east and north-east coasts at high altitudes, yields increased (by +9% and +17% in 2060 and 2090). Overall, water irrigation demand increased by 15 and 24% in 2060 and 2090, respectively, on average across scenarios. It was found that irrigating non-irrigated areas reduced the negative impact of climate change, particularly in high altitudes in the south and north-east. Additionally, the simulation of more drought-resistant varieties was able to limit the negative impact of climate change in areas facing increasing water deficits without changing the irrigation regime. Finally, this study illustrated how areas currently less suitable for sugarcane production, such as high-altitude areas, will become more favorable due to climate change. Adaptation methods, such as changes in irrigation management or the adoption of more drought-tolerant varieties, were identified as effective strategies to cope with climate change and maximize the sugarcane’s potential in a more favorable temperature environment.
-
Section: Archaeology ; Topics: Archaeology
palimpsestR: An R Package for the Identification and Probabilistic Decomposition of Archaeological Palimpsests
10.24072/pcjournal.762 - Peer Community Journal, Volume 6 (2026), article no. e74
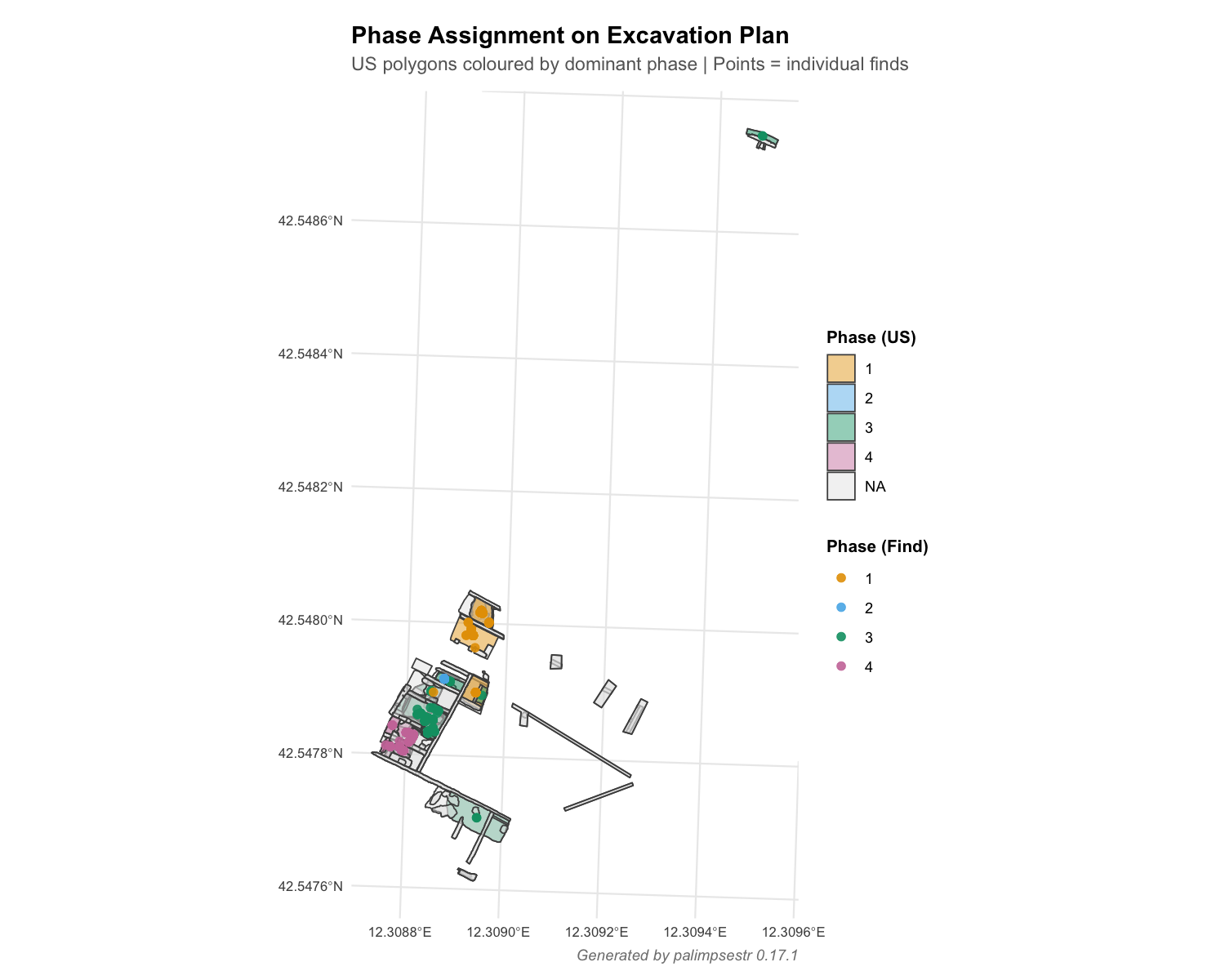
Get full text PDFThis paper presents palimpsestr, an R package and Shiny application dedicated to the identification and probabilistic decomposition of archaeological palimpsests — deposits in which material from multiple occupation phases is superimposed and partially intermixed. Such deposits represent one of the most persistent analytical challenges in field archaeology, and existing tools (Harris Matrix recording systems, k-means spatial clustering, GIS overlays) typically address only one evidence domain at a time. palimpsestr implements the Stratigraphic Entanglement Field (SEF) framework, which integrates four evidence domains — horizontal coordinates, vertical elevation, chronological range, and cultural class — into a single diagonal-covariance Gaussian mixture model fitted by Expectation-Maximisation. The model is augmented with optional taphonomic weighting and stratigraphic entanglement penalties derived from the Harris Matrix. Three interpretable diagnostics — the Stratigraphic Entanglement Index (SEI), Excavation Stratigraphic Energy (ESE), and Palimpsest Dissolution Index (PDI) — allow practitioners to assess deposit coherence, detect potentially redeposited finds, and evaluate the reliability of chronological attribution at the find, unit, and phase levels. The framework supports archaeologists in three principal interpretive tasks: assessing the reliability and coherence of stratigraphic units (distinguishing genuine palimpsests from recording errors); identifying intrusive finds with respect to both their typology and their stratigraphic position; and providing the basis for future statistical estimation of type longevity (the duration of use of pottery and material categories). The package supports CSV/TSV/Excel/SQLite/PostgreSQL data import, GIS export to GeoPackage via the sf package, publication-quality plots via ggplot2 and interactive plots via plotly, and includes a built-in Shiny dashboard for non-programmatic use. Since the original release the statistical core has been substantially refined: the cultural class is now modelled as a per-phase categorical distribution (a Gaussian-times-multinomial mixed-type mixture) rather than as one-hot Gaussian columns, so that stratigraphic units are no longer split across phases by typology; the spatial and vertical similarity kernels are bounded and scale-invariant; the domain weights enter the likelihood and can be cross-validated; a uniform noise component yields a genuine posterior probability of intrusion and shields phase estimates from outliers; and optional treatments propagate per-find dating uncertainty and apply the stratigraphic constraint as a dynamically updated Neighborhood-EM field. The intrusion diagnostic also distinguishes the direction of the chronological mismatch (residual vs. latent-feature), and a companion helper (recommend_setup()) inspects a dataset and reports when its recording resolution limits the achievable inference. We present an overview of the application's functions and demonstrate its use through a case study at the multi-period Roman villa of Poggio Gramignano (Lugnano in Teverina, Italy). We also discuss the methodological assumptions of the SEF framework — in particular the implicit assumption of horizontal stratigraphy and the dependence of phase resolution on the spatial and chronological recording resolution of the data — and outline planned developments.
-
Section: Archaeology ; Topics: Archaeology
Detecting Temporal Relations in Archaeology: Model and Algorithms
10.24072/pcjournal.761 - Peer Community Journal, Volume 6 (2026), article no. e73
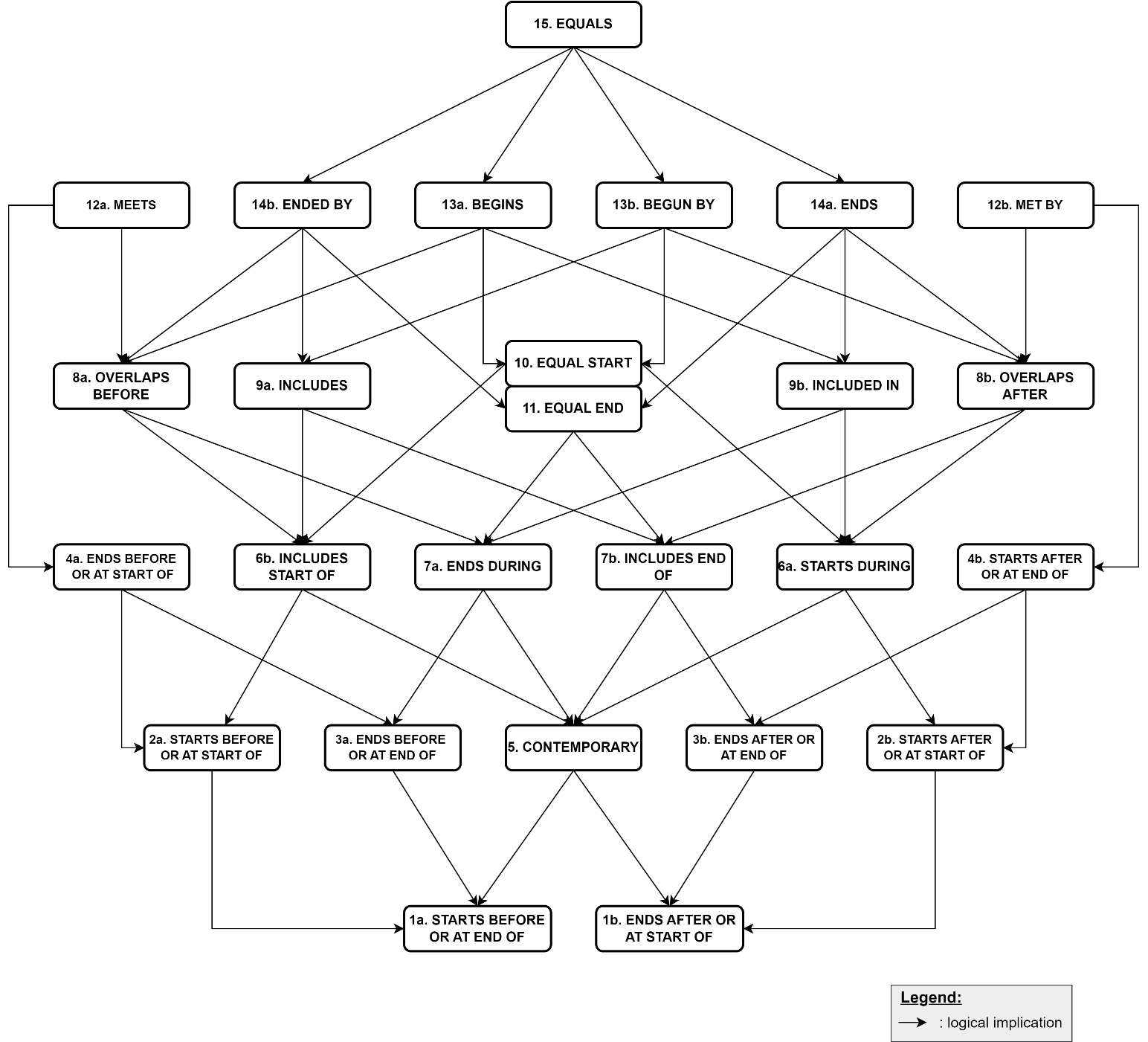
Get full text PDFThis paper addresses the question of finding the optimal temporal relation between two time-periods, in the presence of uncertainties regarding their boundaries (i.e. unknown or bounded start/end). It proposes a fast algorithmic solution, implementated in the Python language, and an archaeological case study related to the Early Bronze Age in the Levant. Open-source Python implementations of our data model and algorithms can be downloaded on the author’s GitHub repository. Our solution is also implemented in the latest version (v. 3) of the ChronoLog modelling tool (https://chrono.ulb.be/).
-
Section: Health & Movement Sciences ; Topics: Physiology, Psychological and cognitive sciences
Mental fatigue impairs cycling endurance performance and perception of effort, but not muscle activation
Souron, Robin;
Sarcher, Aurélie;
Lacourpaille, Lilian;
Boulaouche, Inès;
Richier, Calvin;
Mangin, Thomas;
Gruet, Mathieu;
Doron, Julie;
Jubeau, Marc;
Pageaux, Benjamin
10.24072/pcjournal.760 - Peer Community Journal, Volume 6 (2026), article no. e72
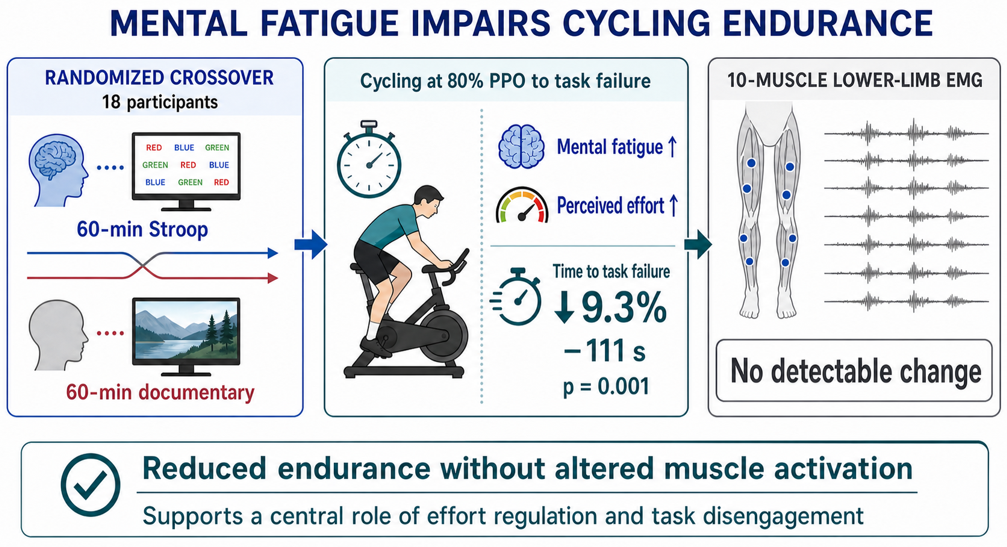
Get full text PDFMental fatigue is induced by prolonged engagement in cognitively demanding tasks and impairs endurance performance. The neuropsychophysiological mechanisms underlying this decreased performance remain unclear, with suggestion that mental fatigue may disrupt motor command and consequently muscle activation. We aimed to test this hypothesis in a repeated cross-over design study in which 18 participants completed two experimental sessions involving a time-to-task failure cycling test at 80% of peak power output. Each cycling task was preceded by 1h of a prolonged Stroop task (Stroop condition) or a neutral control task (Control condition). Mental fatigue was assessed using a visual analog scale anchored with "not fatigued at all" and "extremely fatigued. Perception of effort and surface electromyography from ten lower-limb muscles of the right leg were recorded at regular intervals during cycling. Mental fatigue was higher in the Stroop compared to the Control condition (p = .002). Endurance cycling time was shorter in the Stroop than in the Control condition (887 ± 284 s vs. 999 ± 379 s, respectively; −111 ± 160 s, p = .009). No significant differences in electromyography parameters were observed between Stroop and Control conditions, for any muscle (p > .05). Perception of effort was higher in the Stroop condition from the onset of the cycling task (p = .006), and the rate of increase in perception of effort was significantly higher in the Stroop than Control condition (p = .031). Our findings do not support the hypothesis that mental fatigue alters motor control or increases central motor command, as no changes in muscle activation were detected. Conversely, our results reinforce the notion that prolonged cognitive engagement impairs endurance performance primarily through an increased perception of effort. Future research should consider combining surface electromyography with more sensitive neurophysiological techniques to investigate potential subtle changes in motor drive during dynamic, whole-body tasks under mental fatigue.
Sections
- Animal Science 33
- Archaeology 46
- Ecology 145
- Ecotoxicology & Environmental Chemistry 17
- Evolutionary Biology 111
- Forest & Wood Sciences 10
- Genomics 62
- Health & Movement Sciences 14
- Infections 39
- Mathematical & Computational Biology 32
- Microbiology 22
- Network Science 6
- Nutrition 4
- Neuroscience 13
- Organization Studies 4
- Paleontology 14
- Plants 2
- Psychology 2
- Statistics & Machine learning 1
- Registered Reports 4
- Zoology 29
Membership
Image Credits
The network image was drawn by Martin Grandjean: A force-based network visualization CC BY-SA