
 CC-BY 4.0
CC-BY 4.0
Introduction
RNA-dependent RNA polymerase (RdRp) is a key enzyme in the life cycle of RNA viruses, and serves as the central molecular hallmark of Orthornavirae, providing both a conserved anchor for their identification, and a critical target for functional assessment of genome replication, and evolutionary analysis (Koonin et al., 2015). Its functional core, the palm domain, consists of seven conserved motifs (G, F1–3, A, B, C, D, and E, from the N-terminus to the C-terminus) that fold into a palm-like structure accommodating the RNA template during the replication process (Bruenn, 2003). Among these seven motifs, motifs A, B, and C harbor the conserved residues required for synthesis of the new strand (Tahzima et al., 2025). This conservation is shared among all viral RdRps, with the exception of permuted RdRps, in which motif C is located upstream of motifs A and B without disrupting the overall folded structure of the protein.
Due to its inherent role in viral replication and its ubiquitous presence across major RNA virus lineages, RdRp therefore remains one of the most powerful markers for uncovering viral diversity, especially in the era of metagenomic and metatranscriptomic datasets (Charon et al., 2022; Wolf et al., 2018). The relatively conserved catalytic motifs enable detection even across highly divergent sets of viral clades, making it indispensable for the characterization of the immense “viral dark matter” that remains unculturable and unclassified. By recognizing the central role of RdRp in RNA virus biology, a global community of scientific researchers from diverse disciplines including virology, bioinformatics, structural biology, epidemiology, and evolutionary genomics convened for the inaugural RdRp summit in Valencia, Spain, on 22-23 May 2023 (Charon et al., 2024). The 2023 summit established the foundational need for scalable RNA virus discovery tools and catalyzed the development of community resources such as RdRpCATCH and RdRpScan, providing a clear foundation for the 2025 summit’s subsequent focus on standardization and structure-guided phylogenetics. A second RdRp summit took place in May 2025 in Lisbon, Portugal, with the purpose of discussing the latest advancements, and remaining challenges in the identification, annotation, and phylogenetic interpretation of RdRp sequences. Participants from across the globe gathered in order to share benchmarks, discuss emerging tools and define best practices for large-scale RNA virus discovery. This second summit succeeded in fostering new collaborative initiatives to identify and address the emerging challenges of the field.
Advancements in RdRp Research Since the 2023 Summit
Since the first RdRp summit, several studies have been published in the field of RNA virus discovery and characterization (Figure 1). These studies focus on benchmarking RNA processing methods for High-Throughput Sequencing (HTS) (Schönegger et al., 2023), and on the conceptualization and development of new tools for RdRp identification (Liu et al., 2022) (Figure 2, Top). Technological advances in long-read sequencing have encouraged the publication of studies applying these methods to virome investigations through total RNA sequencing (Pichler et al., 2023). This approach could potentially open new avenues for describing viral diversity at the viral isolate level in the future. More broadly, recent work reflects several emerging trends, including the increasing integration of metatranscriptomic data with ecological and environmental metadata, a shift toward total RNA sequencing approaches to better capture segmented viral genomes, and the growing use of AI-driven homology detection methods to identify highly divergent RdRp sequences. Together, these developments point toward a more comprehensive and data-integrated framework for RNA virus discovery.
Most importantly, progress in the accessibility and scalability of protein structural prediction and modeling has provided the community with powerful tools to investigate the phylogenetic relationships between divergent viruses (Litvin et al., 2025). Widely adopted approaches, including structure prediction tools such as AlphaFold, ESMFold, and ProstT5, together with structure-comparison frameworks like Foldseek, have enabled the detection of deep evolutionary relationships that are often inaccessible through sequence-based methods alone. In parallel, genome completion approaches such as SegFinder are beginning to address challenges associated with fragmented and segmented viral genomes (Heinzinger et al., 2024; Jumper et al., 2021; Lin et al., 2023). These tools also show promise in detecting new clades of RNA viruses with ORFs lacking any detectable homology to known open reading frames (Yin & Fischer, 2006) and completing viral genomes by identifying additional genomic fragments coding for distantly related proteins. These applications are made possible by continuous efforts in the analysis (Chikhi et al., 2025) and annotation (Hou et al., 2024) of global RNAseq databases, which are now available to the RNA virology community.
All these efforts have led to a remarkable expansion of viral taxonomy, both in terms of the number of known viral species and higher-level classifications. Notably, two new phyla have been added to the Riboviria realm: Ambiviricota (Kuhn et al., 2024) and Artimaviricota (Urayama et al., 2024). The discovery of viruses belonging to the phylum Ambiviricota also revealed, for the first time, that RNA viruses can utilize a genomic organization based on circular RNA sequences that replicate via a rolling circle mechanism (Forgia et al., 2023). Accelerated discovery and RdRp-based phylogeny of viruses known as bunyavirals led to the establishment of the class Bunyaviricetes under the Negarnaviricota phylum, accommodating negative-sense RNA viruses.
Modern challenges in RdRp Biology
RNA Virus Categorization
Environmental sampling and sequencing has revolutionized RNA virus taxonomy by expanding the known diversity of RNA viruses (Simmonds et al., 2017), effectively doubling the recognized repertoire over the past five years (twofold increase) through discoveries across a wide range of ecosystems and hosts. Many of these newly identified viruses lack known pathogenicity or clear evolutionary ties to existing taxa (Neri et al., 2022; Wolf et al., 2020). In response to this surge in genomic data, the International Committee on Taxonomy of Viruses (ICTV) has adopted phylogenetic analysis as the main criterion for classifying newly identified viruses, even in the absence of biological information (International Committee on Taxonomy of Viruses Executive Committee, 2020). This shift acknowledges the practical challenges of characterising the rapidly growing number of viral sequences but has also sparked debate among virologists about the implications for species definitions and biological relevance (Gibbs, 2020; Neri et al., 2022; Simmonds et al., 2017; Wolf et al., 2020) (Figure 2, Bottom). This taxonomic framework relies on computational methods that can classify metagenomic sequences despite limited biological information. Reference-based methods are often used for taxonomic classification which compare sequences to curated viral databases, along with marker-based methods, which rely on conserved genes in viral clades. For RNA viruses, the RdRp gene is typically used because it is the most conserved genomic region and is widely used to infer evolutionary relationships (Tang et al., 2022). However, the extensive sequence divergence and structural variability of RdRp often complicate accurate reconstruction and limit taxonomic resolution (Holmes & Duchêne, 2019). To address these challenges, Tang et al. developed RdRpBin, a computational tool combining an alignment-based strategy and machine learning models to improve RdRp sequence detection and classification (Tang et al., 2022). Importantly, RdRpBin represents one example of a broader class of alignment-informed machine learning approaches, rather than a singular solution endorsed by the consortium. Complementary tools, including CHEER and VirHunter, further illustrate the diversity of classification frameworks available, while recent phylogeny-aware methods such as PhyloTUNE and ROADIES extend these capabilities by integrating evolutionary context into sequence analysis (Deng et al., 2025; Gupta et al., 2025).
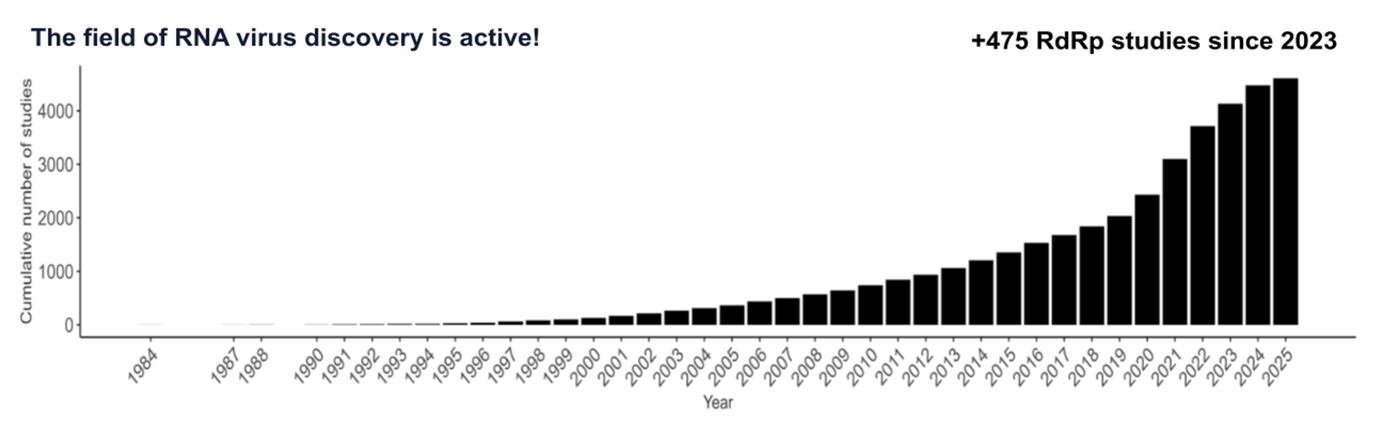
Figure 1 - Cumulative growth of studies focused on RdRp over the years. The rate of new studies and associated SRA submissions appears to be plateauing, whereas the average size of individual sequencing runs is steadily increasing (Chikhi et al., 2025). Since the last RdRp summit in 2023, 475 extra studies on RdRp have been conducted. This figure was generated by doing a keyword search of the SRA for “RdRp”, therefore it may miss some metatranscriptome studies.
A unified RNA virus data landscape
We define the “viral landscape” as the physical, genomic and ecological distribution of viruses in a natural environment, distinguishing it from the “RNA virus data landscape,” which refers to the digital repositories, databases, and bioinformatic frameworks used to store and analyze viral sequences. The analysis of samples using metagenomics methods often reveals diverse and largely unexplored RNA viral genomes in the environment. By advancing the discovery, annotation, and standardized sharing of these viral genomes, the RNA virus data landscape can be unified. Although efforts have been made in the past to standardize the reporting of viral genome data (MIUViG), these were mostly tailored to dsDNA bacteriophages. This framework incorporates RNA virus–specific reporting requirements, including genome strandedness, the presence of subgenomic RNAs, details of RNA extraction and library preparation methods (e.g., poly-A selection versus rRNA depletion), and approaches for recovering segmented genomes. Therefore, a growing need exists for an update to these standards to include the specific needs for RNA virus reporting, e.g. methods used for recovery of segmented genomes, along with user-friendly tools (e.g. SUTVK (Standardized Uncultivated Virus Taxonomy and Knowledge), https://github.com/LanderDC/suvtk) that ensures viral genome data are not only accurately represented but also FAIR-compliant. SUTVK is designed to be compatible with multiple major repositories (including NCBI, ENA, and DDBJ) to facilitate FAIR-compliant data sharing across the global bioinformatics infrastructure. Viral metagenomics is revealing the hidden diversity of viruses across diverse ecological gradients in both urban (e.g., global RNA viromes in cities) (Gao et al., 2024) and natural settings (e.g., rodent-associated viruses in Serbia, water-associated viromes in high-altitude lakes or bee-and pollen-associated viromes in Canadian tree fruit orchards) pointing towards their broader ecological and public health relevance (Vansia et al., 2024; Wu et al., 2025). An integrated vision for building a comprehensive and accessible global viral landscape exists, and is guided through the improvement of methods, tools, and collaborative standards.
Expanding the RNA virus discovery toolkit
Advances in computational biology, sequencing technologies, and machine learning are transforming how RNA viruses are detected, classified, and studied. Traditional alignment-based methods have been supplemented by profile Hidden Markov Models (pHMMs) (https://github.com/dimitris-karapliafis/RdRpCATCH, https://www.biorxiv.org/content/10.64898/2026.02.05.703936v1), structure-aware homology detection, and, more recently, deep learning models that can be applied to identify viruses from genomic and metagenomic data, such as CHEER, VirHunter, Virtifier, and RNN-VirSeeker (Liu et al., 2022; Miao et al., 2022; Shang & Sun, 2021; Sukhorukov et al., 2022). These tools employ convolutional neural networks (CNNs) and recurrent neural networks (RNNs). Despite their versatility, both face limitations in processing biological sequences: CNNs may encounter challenges with inputs of varying lengths and capturing global correlations, while RNNs struggle with longer sequences due to vanishing or exploding gradients and difficulties in capturing long-term dependencies. To address this, Hou et al. employed a new AI model based on transformer architecture (i.e., LucaProt) for RNA virus discovery that utilizes both protein sequences and the structural characteristics of viral RdRp sequences, and shows its power for uncovering remote viral RdRp homologies and functional signatures, even in sequences with no detectable similarity to known references (Hou et al., 2024; Nakagawa & Sakaguchi, 2024).
These tools enable researchers to uncover RNA viruses that would have previously escaped detection due to their low sequence similarity to known taxa. At the same time, long-read and single-cell sequencing technologies are enhancing genome assembly and host linkage, especially in complex environmental samples. This growing arsenal of methods facilitates the discovery of the hidden RNA virosphere, annotation of functional viral elements, and inference of host–virus interactions, thereby enriching both taxonomic resolution and biological interpretation.
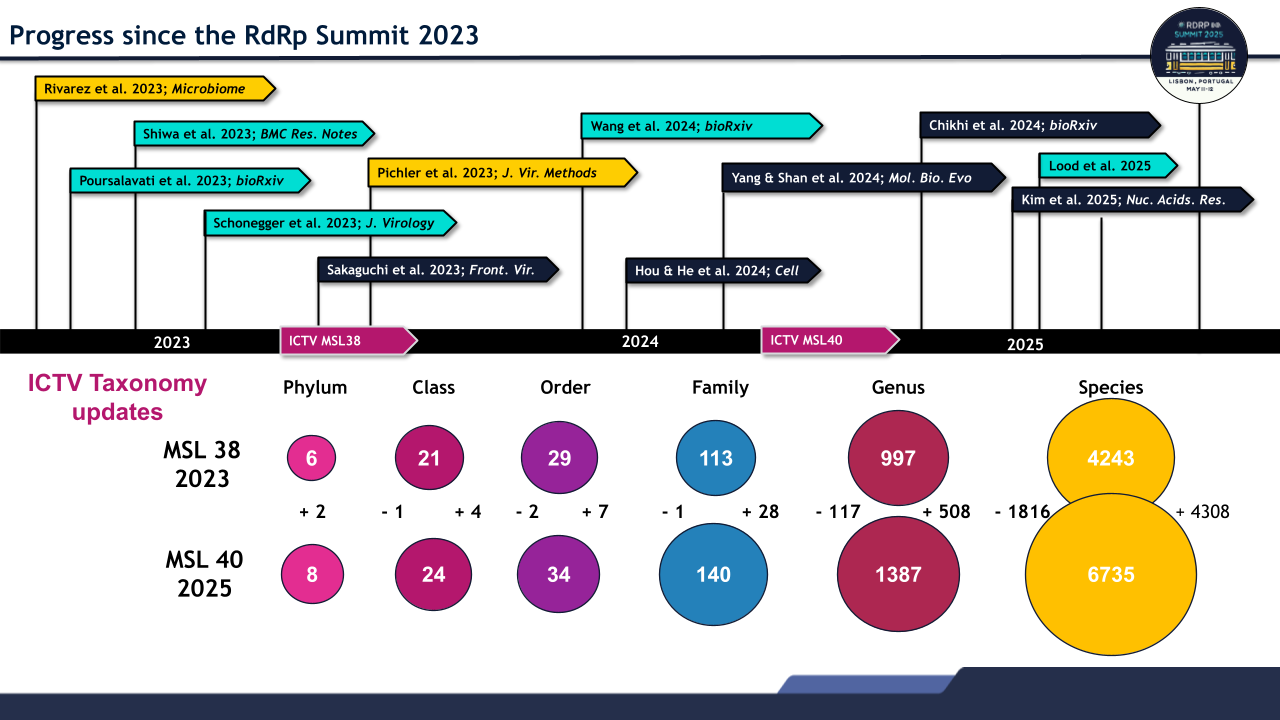
Figure 2 - Advances in RNA virus biology and ICTV growth since 2023. (Top) Progress in RNA virus research following the inaugural RdRp Summit in 2023 includes long-read and single-cell sequencing, structure-based phylogenetics, large-scale protein modeling (AlphaFold, ESMfold, Foldseek, BFVD), and AI-driven discovery tools (LucaProt, CHEER, VirHunter), which together have revealed deeply divergent RNA virus lineages and illuminated the hidden virosphere. (Bottom) Each of the rings illustrates the rapid expansion of the ICTV taxonomy, including new phyla (Ambiviricota and Artimaviricota).
Mining the planetary virosphere at scale and depth
Several global initiatives, such as VIRION (https://www.viralemergence.org/virion), PREDICT (https://ohi.vetmed.ucdavis.edu/programs-projects/predict-project), and the NIH Human Virome Program (https://commonfund.nih.gov/humanvirome) have been instrumental in elucidating viromes across different environments, organisms, or ecosystems, each implementing distinct sampling and analytical strategies (Carlson et al., 2022; Wallace et al., 2025; Wang et al., 2025; Wu & Peng, 2024). Alongside these studies are the increasing number of research practices which introduce challenges in verifying the identity and taxonomy of uncultivated viruses. This underscores the need for standardized methodologies that would ensure interoperability, reproducibility, and robust results that are both verifiable and reliable.
Leveraging large-scale computational and evolutionary approaches allows for the discovery of hidden dimensions of the planet’s virosphere and enables the assessment of emerging viral threats at unprecedented scales (Chikhi et al., 2025; Edgar et al., 2022). For example, the Logan project (https://logan-search.org) provides a framework for the assembly and analysis of vast amounts of complex and fragmented genomic data. While recent breakthroughs have been driven largely by advances in computational methods, the standardized methodologies advocated by the consortium must extend across the entire pipeline, encompassing ecological sampling, RNA extraction, sequencing protocols, and downstream bioinformatic processing. This end-to-end standardization is essential to ensure that insights derived from large-scale analyses are comparable, reproducible, and biologically meaningful. The identification of endogenous viral elements embedded in public databases, using uncharacterized proteins as a window into ancient viral integrations, provides us with a deeper understanding of their evolutionary legacies in host genomes and their biological implications (Brown & Firth, 2024). The integration of bioinformatics, evolutionary biology, and big data illuminates the origins, trajectories, and zoonotic potential of RNA viruses on a global scale. Taken together, these tools provide insights into viral emergence, biological history, and future threats to human and animal health.
Illuminating the hidden RNA virosphere
Approaches to uncovering the vast hidden diversity of RNA viruses, often referred to as “viral dark matter”, offer insights into one of the largest biological diversity reservoirs on the planet. By harnessing artificial intelligence (AI) to systematically document the hidden RNA virosphere, we have begun to understand and demonstrate how machine learning can be applied to detect and classify novel RNA viruses from increasingly complex datasets (Hou et al., 2024). Structural bioinformatics methods (BFVD-Foldseek) use protein-folding and alignment to identify deeply divergent viral sequences that elude current conventional sequence homology-based detection methods and further illuminate the structural and functional diversity (and their relationship) of viral dark matter (Kim et al., 2025). The extreme diversity presented by the marker gene RdRp can be examined using deep evolutionary analysis to demonstrate how this core viral enzyme varies significantly across lineages and diverse environments. The potentially transformative synergy between advances in AI methods, structural biology, and evolutionary sequence-based modeling is poised to fundamentally reshape our understanding of the breadth and depth of RNA viral diversity. The deliberate harmonization of these approaches constitutes a major strength of the field and has the potential to exert a lasting impact on future discoveries. Achieving this harmonization will require coordinated community efforts, including the establishment of benchmarking datasets for method evaluation, the adoption of standardized metadata reporting frameworks and the integration of structure-informed annotations and predictions into formal ICTV taxonomic workflows. Harmonization will be achieved through the establishment of community-agreed benchmarking datasets, the adoption of standardized metadata reporting, and the integration of structural predictions into official ICTV taxonomic pipelines. Together, these steps will enable more robust, reproducible, and scalable frameworks for RNA virus discovery and classification.
Towards community-driven solutions and future initiatives
The RdRp Summit 2025 brought together researchers working on current challenges of RNA virus research, but also in actively addressing them through community-driven collaboration. As the field of RNA biology continues to expand at an unprecedented pace and is spurred on by advances in short- and long-read sequencing technologies, structural prediction, AI, and metagenomics - it has become increasingly important to align efforts around shared methodologies, standards, and infrastructures (Zielezinski et al., 2025).
A key goal of the summit is to foster a community that remains active between the biannual meetings. To support this objective, the summit serves as a hub to connect researchers facing similar challenges, enabling them to work collectively toward shared solutions. To this end, participants were invited to propose ideas for community-driven initiatives. A dedicated session was held to form collaborative groups around selected topics of mutual interest. In this report, we present the community projects that will be supported and developed by the RdRp Summit over the next two years (Figure 3). We propose three prioritized and interdependent initiatives: (1) the establishment of foundational Benchmarking Challenges to rigorously evaluate and standardize computational tools, (2) the application of these validated tools to Virus–Host Relationship Inference, and (3) the integration of these insights into Structure-Guided Phylogeny.
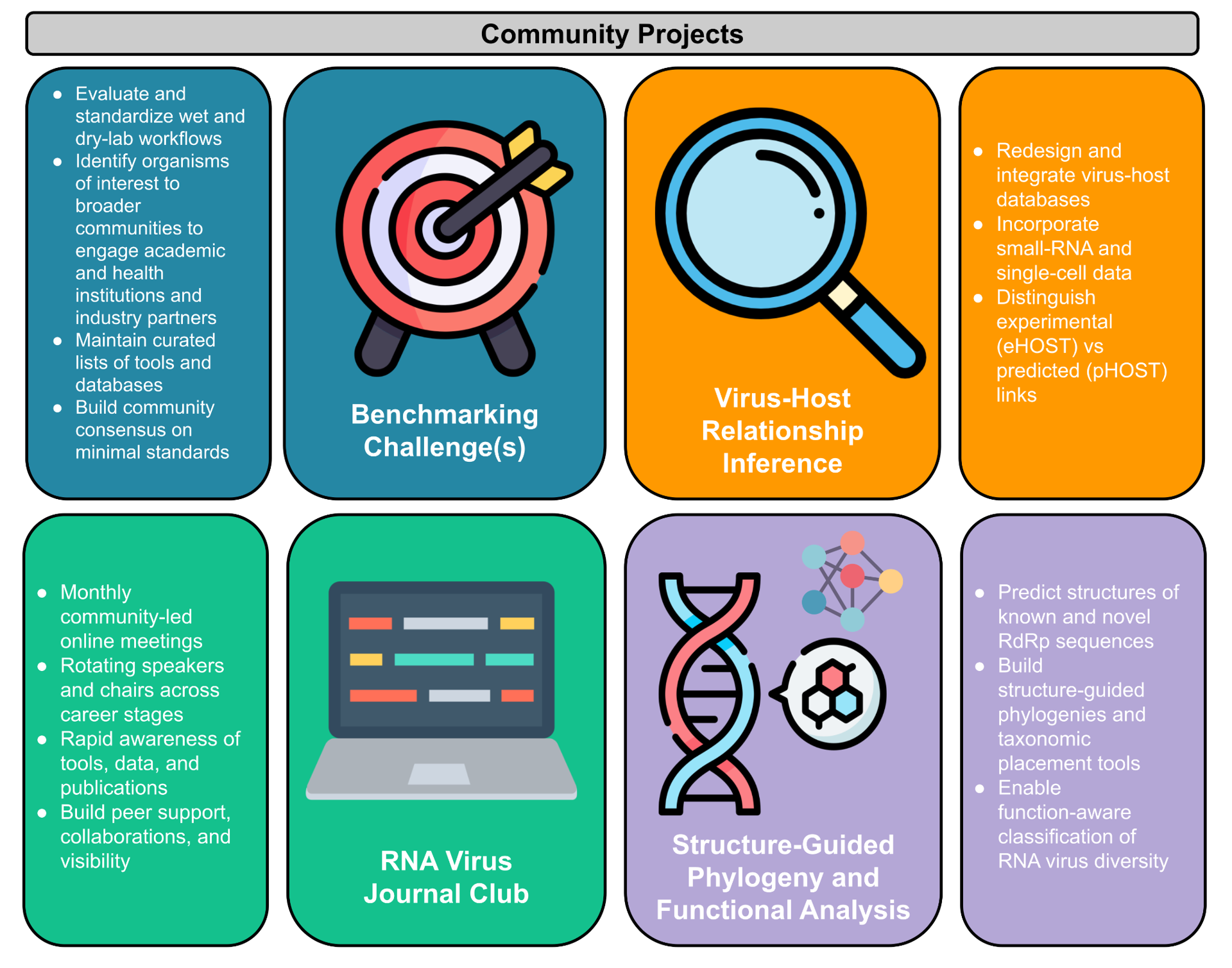
Figure 3 - Community-driven initiatives launched at the second RdRp Summit. Four community projects were proposed to address critical gaps in RNA virus research identified by participants of the RdRp Summit 2025. (1) Benchmarking Challenges aim to develop gold-standard datasets, metrics, and curated tool lists to standardize both experimental and computational workflows. (2) Virus-Host Relationship Inference focuses on improving host assignment through integrated databases, multi-layered prediction approaches, and hackathon-driven curation. (3) RNA Virus Journal Club provides a platform for regular knowledge exchange, collaborative discussions, and community building. (4) Structure-Guided Phylogeny and Functional Analysis seeks to classify deeply divergent RNA viruses by integrating structural prediction, comparative analyses, and phylogenetic frameworks. Together, these initiatives establish the foundation for coordinated, sustained progress in RNA virus discovery.
Community projects
To meet this need, the summit aims to establish collaborative initiatives that have been proposed and launched by the community. For example, as part of the 1st RdRp Summit, the community project of RdRpCATCH -RdRp Collaborative Analysis Tools with Collections of pHMMs- was introduced (https://github.com/dimitris-karapliafis/RdRpCATCH). Notably, the first RdRp Summit demonstrated that coordinated community action can substantially accelerate tool validation and comparative assessment, providing a scalable framework for benchmarking emerging methodologies across diverse datasets. This project consolidated multiple RdRp pHMM databases developed over the past five years into a single resource, addressing fragmentation in the field and reducing the technical barriers to the discovery of RNA viruses. The 2nd RdRp Summit led to the launch of multiple community projects, each designed to tackle key gaps in RNA virus discovery.
Benchmarking Challenge(s)
The “Benchmarking Challenge” represents a coordinated community effort to establish standardized frameworks for the evaluation of RNA virus discovery and assembly tools. Central to this initiative is the development of shared benchmarking datasets spanning synthetic constructs, mock communities, and real-world multi-virome samples, enabling consistent and reproducible performance assessment across methods. By defining common evaluation metrics and testing conditions, this effort seeks to ensure comparability between tools, reduce fragmentation in methodological development, and prevent the proliferation of unvalidated approaches. Ultimately, the Benchmarking Challenge provides a foundation for rigorous, transparent, and scalable tool validation, supporting more reliable discovery and characterization of RNA viral diversity. The benchmarking challenges working group highlighted several core issues that are considered important. These include securing sustainable funding, defining the scope of the work, including which viruses to prioritize, and ensuring long-term oversight, since meaningful benchmarking is likely to be a slow and iterative process. In order to provide gold standard data sets, the objectives of this working group include identifying what can and should be benchmarked across both wet-lab experimental protocols and dry-lab computational analysis, including the generation of synthetic datasets, and determining how these benchmarks can be effectively implemented and compared in the field. To strengthen our approach, the group also emphasized learning from related initiatives to generate gold standard benchmarking data sets, such as existing certification and standards projects (i.e. CAMDA, https://bipress.boku.ac.at/camda2025) and CASP (Critical Assessment of Structure Prediction, https://predictioncenter.org/index.cgi), which could provide useful models and prevent duplication of effort. Additionally, the group will maintain a curated list of software, tools, databases, and resources for RNA virus analysis, prediction, annotation, phylogenetics, and related research, which is available at: https://github.com/rdrp-summit/awesome-rna-virus-tools.
Virus-Host Relationship Inference
We suggest that databases being redesigned for virus–host inference should prioritize the integration of host–virus association metadata with genomic features (e.g., k-mer profiles, GC content), rather than duplicating the storage of raw sequencing data, which remains the responsibility of primary repositories such as the Sequence Read Archive. Participants identified key priorities including the redesign of database curation and integration, where current resources were deemed inadequate. Current collaborative efforts include building more accurate web-curated databases supported by both manual and semi-automated approaches. Databases will be extended by employing multiple dataset-specific statistics (e.g., k-mer analysis, GC-content, nucleotide frequencies), and expanding to underutilized data types such as small RNA sequencing and single-cell RNA data. We also emphasize distinguishing experimental “eHOSTs” from predicted host assignments “pHOSTs”, tracking co-occurrence in raw data and integrating evolutionary features like mutation and recombination rates. It is also important to distinguish between hosts as the organism(s) within whose cells a virus replicates, and the organism from which virus might be collected from, as this can lead to spurious virus-host relationships without evidence that the virus successfully replicates within a particular organism.
The organization and importance of hackathons, community-driven curation, and benchmarking efforts will be central in order to make progress on the massive manual curation task. Broader considerations include the role of host taxonomic rank in classification and its importance, and the integration of viral-like elements and host-protein interactions, as well as the need for standardization of approaches. The group is currently working and will produce a comprehensive review of AI-based virus-host prediction tools, highlighting the necessity of multi-layered computational prediction methods and statistical frameworks, and community coordination.
RNA Virus Journal Club
To facilitate communication among RNA virus researchers, the RNA Virus Journal Club was established. Organized by the second RdRp Summit participants, the Journal Club serves as a platform to share the latest updates in the field of RNA virus research. Importantly, monthly meetings have rotating chairs and times to promote inclusivity. Within online meetings, the community could exchange research insights and challenges, get help and advice from peers, and find potential collaborators. Each session includes a presentation from one main speaker, followed by open discussion. Community members of all career stages are encouraged to nominate speakers and chairs and participate actively, ensuring the meetings reflect the needs and interests of the community. Short summaries of the meetings are available afterwards, also promoting offline discussion. The participation and speaker nomination forms are available at the RdRp summit website (https://rdrp.io). The Journal Club started in September 2025, and we hope this will be a useful tool for the community to keep active networking. Since its inception in September 2025, the journal club has maintained strong engagement, with an average attendance of 40-60 researchers per session, representing a diverse, global cross-section of the community.
Structure-Guided Phylogeny
The immense diversity of viral proteins results in a scarcity of sequence homologs in existing databases, posing major challenges for protein comparison and annotation (Kuchibhatla et al., 2014; Terzian et al., 2021). However, advances in highly accurate and scalable tools for protein structure prediction AlphaFold, ESMfold, ProstT5 (Heinzinger et al., 2024; Jumper et al., 2021; Lin et al., 2023) and comparison (Foldseek) (van Kempen et al., 2024), combined with improved understanding of the statistical properties of unrelated structural folds, now enable the detection of homologs with near undetectable sequence similarity.
By comprehensively predicting the structures of the known RdRp diversity we can expand our search space for detecting novel distant homologs; not only relying on amino acid similarity or motif conservation but also utilising the deeper conservation present on the structural level of the proteins. The Big Fantastic Virus Database (BFVD, https://bfvd.foldseek.com/) (Kim et al., 2025) has already provided a great starting point for enabling structure-based homology searches against virus protein structures. However, there is a growing number of distant RdRp sequences whose structure has not been predicted yet. This unexplored diversity is exemplified by the findings of the LucaProt approach, describing 180 RNA virus supergroups based on deep RdRp homology, 23 of which were previously unknown and only 21 of which have been classified taxonomically (Hou et al., 2024).
The already known RdRp sequence diversity can largely help with confidently predicting their corresponding structures, since many of the best performing prediction methods rely on comprehensive multiple sequence alignments (MSA). Combining the LucaProt RdRp dataset with other expansive sequence databases (e.g., Logan) may improve MSA-based predictions. For cases where close known homologs remain sparse, protein Language Models (pLM)-based predictions can produce improved structures, having been shown to perform better than MSA-based methods when alignments are shallow (Mifsud et al., 2024).
A database of RdRp structures (both predicted and experimentally determined) can aid in the quest to uncover more of the hidden RNA virosphere but also perform structure-guided phylogenetics of the RNA virosphere. Given the exponential increase in available (predicted) protein structures, new methods have been developed for structure-based sequence alignment (Gilchrist et al., 2024) and phylogenetics (Puente-Lelievre et al., 2024). We believe that these approaches can provide higher resolution in the more ancient parts of the RdRp evolutionary history that remain unresolved; for example, delineating the relatedness of ‘orphan’ taxonomic groups. We envision the creation of a comprehensive RdRp structure database, datasets of structure-guided phylogenies for all uncovered RdRp diversity, and a tool for placing novel RdRp candidates into this diversity based on both sequence and structural homology.
Future initiatives
Quantifying evolutionary novelty
The discovery of novel RdRps at scale has been facilitated by the growing availability of diverse and often underexplored sequencing datasets, including metatranscriptomes, viromes (enriched or rRNA-depleted), dsRNA sequencing approaches, and specialized protocols such as fragmented and loop primer ligated dsRNA sequencing (FLDS), many of which are publicly accessible through repositories such as the Sequence Read Archive. Within this landscape, total RNA sequencing and poly(A)-selected sequencing represent complementary methodological strategies: total RNA approaches enable the capture of segmented and non-polyadenylated viruses, while poly(A) selection enriches for eukaryotic host transcripts and specific viral clades. Coupled with increasingly sensitive homology detection methods, these advances have substantially expanded our ability to detect and characterize RNA viruses across diverse environments. Many (if not most) RNA virus discovery studies typically report numbers of newly discovered RdRp operational taxonomic units, OTUs (groups of homologous sequences clustering at an arbitrary identity threshold), or similar terms that communicate the scale but not depth of discovery. For example, a now classic study by (Li et al., 2015) reported the discovery of 112 novel negative sense single-stranded RNA viruses in arthropods but at the time did not have the quantitative means (e.g., branch length–based measures across large datasets). to communicate that a considerable number of these viruses comprised what is a very distinct family-level group now referred to as chuviruses.
At the RdRp Summit in Lisbon in May 2025, a metric was proposed that could help turn metatranscriptomic RNA virus discovery studies more quantitative by adapting what is called “phylogenetic diversity” in ecology. Branch lengths in phylogenetic trees typically represent independent amounts of character evolution (e.g. amino acids) and the sum of branch lengths added to trees by new sequences can be thought of as its evolutionary novelty. When sequences detected in a sample are analysed in this way, statements like “we detected 2 viruses (1 novel and 1 previously described)” can be refined further into “we detected 2 viruses contributing a total of 0.64 aa/subs/site and 0.0029 aa/s/s”, imparting to the reader that one sequence is substantially novel while the other is very closely related to known ones. While phylogenetic diversity metrics provide a powerful framework for quantifying viral discovery, their application is currently constrained by alignment quality and phylogenetic depth. Highly divergent viruses that cannot be reliably aligned remain difficult to incorporate into such analyses. Future iterations of this approach may therefore benefit from integrating alignment-free measures of evolutionary distance, including structure-based comparisons derived from tools such as Foldseek.
RNA viruses lend themselves naturally to such studies on account of the RdRp being a conserved gene shared by all RNA viruses. Furthermore, phylogenetic diversity can be quantified on phylogenetic trees encompassing theoretically, as few as two most closely related sequences, minimising phylogenetic uncertainty arising from poor alignment quality. One implementation of phylogenetic diversity quantification was used on orthomyxoviruses in (Batson et al., 2021), showcasing how phylogenetic diversity can: i) track the success of discovery methods to date, ii) visually track where novelty is found across a phylogenetic tree (using a method adapted from (Obbard, 2018) forecast saturation (or not) of novelty, and iv) identify virus sub-groups contributing most novelty. Going forward, the quantitative tracking of RNA virus discovery efforts could be done at the initiative of study authors themselves or by other groups on publicly available data, with additional considerations such as focusing on novelty contributed by specific virus sub-groups (e.g. influenza vs quaranjaviruses) or host groups (e.g. arthropods vs vertebrates).
Recovering complete segmented genomes
For many unsegmented RNA viruses, modern metatranscriptomic methods suffice: a long open reading frame found on the same contig as known viral genes can usually be accepted as viral. In contrast, segmented RNA viruses - especially those with numerous short segments such as reoviruses or orthomyxoviruses - pose greater challenges. Their smaller segments often lose detectable amino acid homology faster than others (e.g. RdRp-encoding segments), and without physical linkage to known viral genes, they can vanish into metagenomic “dark matter,” leaving genomes incomplete.
Previously, small interfering RNA (siRNA) sequencing in arthropods helped address this problem by identifying dsRNA-derived sequences, enabling co-occurrence-based reconstruction of segmented genomes (Webster et al., 2015). This approach led to the discovery of Galbūt virus, a common Drosophila melanogaster partitivirus. Co-occurrence-based reconstruction has also proven effective in metatranscriptomic studies of individual hosts, revealing, for instance, that four mosquito-associated quaranjaviruses (Orthomyxoviridae) possess eight-segment genomes, with the smallest three lacking any recognisable homology (Batson et al., 2021). Falling sequencing costs and tools such as SegFinder (Liu et al., 2025) arelikely to make complete genome reconstruction more routine. Ideally, though, researchers should take the initiative to explore and report RNA virus diversity in full (i.e. genome-resolved characterization of RNA viruses that includes complete genome reconstruction, linkage of all segments in multipartite viruses, and comprehensive analysis of sequence, structure, function, and evolutionary relationships beyond single-marker genes such as RdRp).
Conclusion
By building on progress since the inaugural meeting in 2023, participants demonstrated breakthroughs in sequencing, structural modeling, and advances in AI-driven methods for characterization, which are quickly expanding viral taxonomy and illuminating the vast hidden dark matter of the RNA virus world. The summit has identified a number of pressing challenges in our field: this includes the need for standardized annotation, data sharing and advancing structure-guided phylogenetics. The summit also launched a number of community-driven initiatives: such as benchmarking challenges, virus-host inference projects and an RNA virus journal club, which can be attended by anyone. Through coordinating efforts across multiple disciplines, the summit has laid the groundwork for a shared research initiative that will accelerate the discovery, improve the classification of, and provide deeper evolutionary insights into the global RNA viral ecosystem. Join our initiative through https://rdrp.io/, which includes contact information and invitations to our communication channels.
Acknowledgements
The authors gratefully acknowledge the contributions of all participants whose collaboration, presentations, and shared expertise made this consensus statement possible. Their collective efforts and commitment to the RNA virus field were essential. We acknowledge Valentyn Bezshapkin (https://orcid.org/0000-0002-0912-4371) for their insightful feedback and assistance. Preprint version 5 of this article has been peer-reviewed and recommended by Peer Community In Microbiology, (https://doi.org/10.24072/pci.microbiol.100236; Massart, 2026).
Data, scripts, code, and supplementary information availability
Not applicable.
Conflicts of interest disclosure
The authors declare they comply with the PCI rule of having no financial conflicts of interest. The 2025 RdRp summit was supported by the Peer Community In (PCI) open-science initiative
Funding
The 2025 RdRp summit was supported by the Peer Community In (PCI) open-science initiative (https://peercommunityin.org) and the International Society for Microbial Ecology (ISME). RR was supported by funding provided by the US National Science Foundation (NSF DBI 2515340) to the Viral Emergence Research Institute (https://www.viralemergence.org). GD is supported by EMBO installation grant EMBO-IG-5305-2023. TD was supported by grants from the Research Council of Finland (330977) and the Kone Foundation. LDC was supported by the Research Foundation Flanders (11L1325N).